Clear Sky Science · sv
UBQLN2 kopplar proteotoxisk stress till lipidmetabolism vid neurodegeneration
Hur nervceller får slut på bränsle
Varför dör vissa nervceller i sjukdomar som amyotrofisk lateralskleros (ALS) och frontotemporal demens (FTD)? Denna studie visar att ett enda cellulärt "trafikpolis"-protein hjälper till att koppla samman två viktiga uppgifter i våra neuroner: att rensa skadade proteiner och att hantera fetter och kolesterol. När denna trafikpolis inte fungerar rätt börjar nervceller förbruka sina fettreserver på ett skadligt sätt, förlorar viktiga byggstenar för membran och dör till slut. Att förstå denna dolda energikris kan peka mot nya sätt att hålla sårbara hjärnceller vid liv längre.
En bruten länk mellan städning och energi
Friska hjärnceller återvinner ständigt uttjänta proteiner och anpassar hur de använder socker och fett för att möta sina energibehov. Proteinet UBQLN2 fungerar som en transportör som styr skadade eller överflödiga proteiner till cellens avfallsmekanismer. Familjer med vissa UBQLN2‑mutationer kan utveckla ALS och FTD, vilket antyder att fel i detta städsystem kan bidra till sjukdomsutveckling. Författarna använde motorneuroner härledda från mänskliga stamceller med patientliknande UBQLN2‑mutationer, tillsammans med hjärnorganoider och musmodeller, för att se hur detta protein påverkar balansen mellan protein‑kvalitetskontroll och energimetabolism.
När glukos blir knapp, spårar fettnedbrytningen ur
Eftersom ALS‑ och FTD‑hjärnor ofta visar nedsatt glukosanvändning stressade teamet sina motorneuroner genom att ta bort glukos, vilket tvingade cellerna att förlita sig mer på fett. Med en kombination av proteomik, lipidomik och RNA‑sekvensering fann de att UBQLN2‑mutationer saktar ner omsättningen av tusentals proteiner och starkt förändrar metabolismrelaterade vägar. Vid energistress ökade de mutanta neuronerna långtids‑oxidation av långkedjiga fettsyror i mitokondrierna och visade en påtaglig förlust av lipidkroppar—små fettreserver—och kolesterolrika lipider. Synaptiska vesiklar, de små säckar som släpper ut kemiska signaler mellan nervceller, blev sköra, och neuronsöverlevnaden sjönk. Att tillsätta måttliga mängder kolesterol återställde delvis vesiklarnas integritet och skyddade mutanta neuroner och hjärnorganoider, vilket lyfter fram kolesterolförlust som en nyckeldel av problemet.
Två enzymer tippar balansen
Vid en närmare granskning frågade forskarna vilka specifika proteiner UBQLN2 normalt kontrollerar för att hålla lipidmetabolismen i schack. De identifierade två enzymer, ILVBL och ALDH3A2, som hjälper till att omvandla vissa fettmolekyler till former som kan förbrännas i mitokondrierna. I friska celler binder UBQLN2 dessa enzymer och styr dem mot nedbrytning, särskilt under stress, vilket begränsar hur aggressivt fetter matas in i mitokondrierna. I UBQLN2‑mutanta neuroner däremot blir ILVBL och ALDH3A2 kvar längre och ackumuleras vid kontaktställen mellan lipidkroppar och mitokondrier. Detta driver överdriven fettförbränning, aktiverar en energikännande väg som dämpar kolesterolproduktionen och dränerar de lipider som behövs för att upprätthålla robusta neurala membran och synapser.
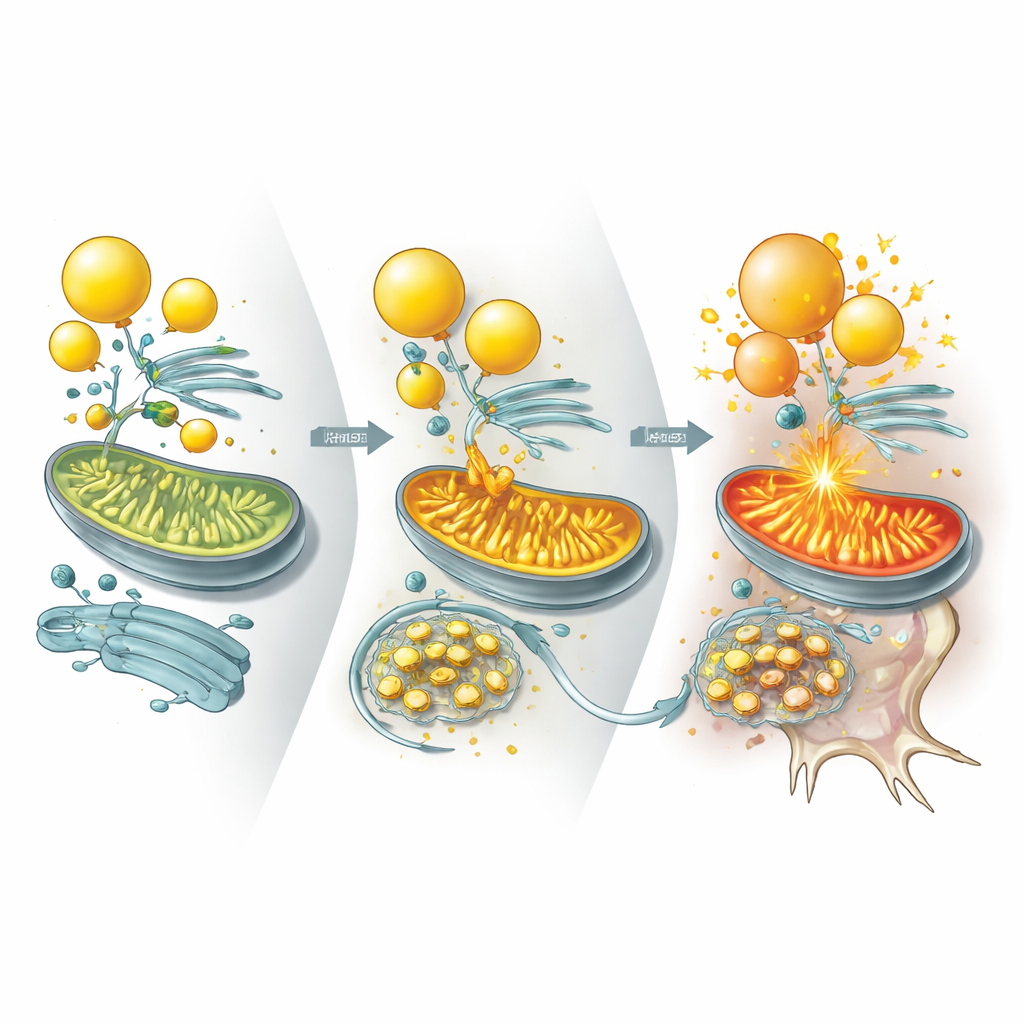
Bevis från mini‑hjärnor, möss och patientvävnader
Samma mönster framträdde i flera system. UBQLN2‑mutanta hjärnorganoider utvecklade färre lipidkroppar och visade mer neuronsdöd vid energistress, vilket återigen kunde mildras med kolesteroltillskott. I möss konstruerade för att uttrycka en sjukdomsassocierad UBQLN2‑variant ackumulerades ILVBL och ALDH3A2 i neuronerna, lipidkroppar försvann och degeneration uppstod, vilket ledde till motoriska och minnessvårigheter. Viktigt är att sänkning av ILVBL‑ eller ALDH3A2‑nivåer i dessa möss återställde lipidlagren, förbättrade neuronsöverlevnad och delvis räddade rörelseförmågan. Författarna undersökte också mänsklig ryggmärgsvävnad från patienter med sporadisk ALS präglad av TDP‑43‑proteinaggregat. I dessa prover fastnade UBQLN2 i TDP‑43‑inklusioner, ILVBL och ALDH3A2 hopade sig, lipidkroppar var uttömda och kolesterolrelaterade lipider var reducerade—en återspegling av vad som observerats i de experimentella modellerna.
Att vända en ond cirkel till ett mål
För icke‑specialister är huvudbudskapet att vissa former av ALS och FTD kanske inte bara handlar om giftiga proteinkluster utan också om en långsam kollaps av neuronernas bränsle‑ och membranekonomi. UBQLN2 håller normalt lipid‑bearbetande enzymer i kort koppel så att fettförbränningen stöder snarare än saboterar cellhälsan. När UBQLN2 är muterat eller fångas upp av TDP‑43‑aggregat löper dessa enzymer fritt, lipidkropparna krymper, kolesterolproduktionen sjunker och synapser försvagas—en ond cirkel som slutar i neurodegeneration. Genom att peka ut denna UBQLN2–ILVBL/ALDH3A2‑axel, och visa att antingen att dämpa dessa enzymer eller återställa kolesterol kan vara skyddande, öppnar studien nya terapeutiska vinklar för att sakta eller förebygga förlust av nervceller i ALS, FTD och närliggande sjukdomar.
Citering: Liu, Y., Huang, Z., Hsu, YW. et al. UBQLN2 links proteotoxicity with lipid metabolism in neurodegeneration. Nat Neurosci 29, 782–795 (2026). https://doi.org/10.1038/s41593-026-02226-y
Nyckelord: ALS, frontotemporal demens, lipidmetabolism, proteinhomostas, neurodegeneration