Clear Sky Science · it
UBQLN2 collega la proteotossicità al metabolismo lipidico nella neurodegenerazione
Come i neuroni restano senza carburante
Perché alcuni neuroni muoiono in malattie come la sclerosi laterale amiotrofica (SLA) e la demenza frontotemporale (FTD)? Questo studio mostra che una singola proteina cellulare, che funge da «vigile del traffico», collega due funzioni vitali nei nostri neuroni: eliminare le proteine danneggiate e gestire grassi e colesterolo. Quando questo vigile non funziona correttamente, i neuroni consumano le loro riserve lipidiche in modo dannoso, perdono componenti chiave per la costruzione delle membrane e infine muoiono. Comprendere questa crisi energetica nascosta potrebbe indicare nuove strade per mantenere in vita più a lungo le cellule cerebrali vulnerabili.
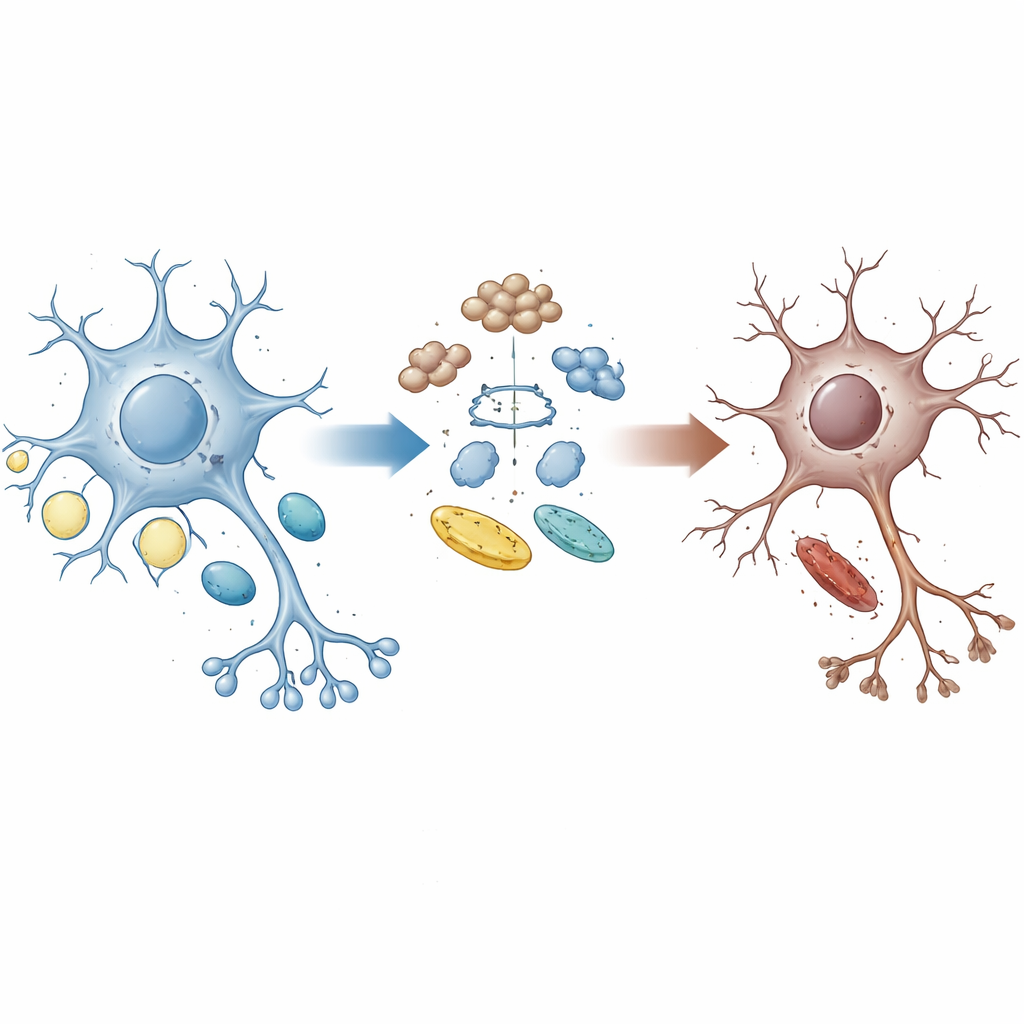
Un collegamento interrotto tra pulizia e energia
Le cellule cerebrali sane riciclano costantemente le proteine usurate e regolano l’uso di zuccheri e grassi per soddisfare il loro fabbisogno energetico. La proteina UBQLN2 agisce come uno shuttle che indirizza le proteine danneggiate o non necessarie verso i sistemi di smaltimento della cellula. Famiglie con determinate mutazioni di UBQLN2 possono sviluppare SLA e FTD, suggerendo che il malfunzionamento di questo sistema di pulizia possa contribuire alla malattia. Gli autori hanno utilizzato motoneuroni derivati da cellule staminali umane portatori di mutazioni UBQLN2 simili a quelle dei pazienti, insieme ad organoidi cerebrali e modelli murini, per valutare come questa proteina influenzi l’equilibrio tra controllo di qualità delle proteine e metabolismo energetico.
Quando il glucosio scarseggia, la combustione dei grassi va fuori controllo
Poiché i cervelli affetti da SLA e FTD mostrano spesso un uso inefficiente del glucosio, il gruppo ha messo sotto stress i motoneuroni rimuovendo il glucosio, costringendo le cellule a fare maggiore affidamento sui grassi. Con una combinazione di proteomica, lipidomica e sequenziamento dell’RNA, hanno scoperto che le mutazioni di UBQLN2 rallentano il turnover di migliaia di proteine e alterano in modo marcato le vie metaboliche. Sotto stress energetico, i neuroni mutanti aumentavano l’ossidazione degli acidi grassi a catena lunga nei mitocondri e mostravano una netta perdita di goccioline lipidiche — minuscoli depositi di grasso — e di lipidi ricchi di colesterolo. Le vescicole sinaptiche, i piccoli sacchi che rilasciano segnali chimici tra i neuroni, divennero fragili e la sopravvivenza neuronale diminuì. Aggiungere quantità moderate di colesterolo contribuì a ripristinare l’integrità delle vescicole e a proteggere neuroni mutanti e organoidi cerebrali, evidenziando la perdita di colesterolo come elemento chiave del problema.
Due enzimi squilibrano il sistema
Approfondendo, i ricercatori hanno chiesto quali proteine specifiche UBQLN2 controlli normalmente per tenere a freno il metabolismo lipidico. Hanno identificato due enzimi, ILVBL e ALDH3A2, che aiutano a convertire certi composti grassi in forme pronte per essere bruciate nei mitocondri. Nelle cellule sane, UBQLN2 si lega a questi enzimi e ne indirizza la degradazione, soprattutto durante lo stress, limitando quanto energicamente i grassi vengano inviati ai mitocondri. Nei neuroni con mutazioni di UBQLN2, invece, ILVBL e ALDH3A2 permangono più a lungo e si accumulano nei siti di contatto tra goccioline lipidiche e mitocondri. Questo spinge a un eccesso di combustione lipidica, attiva una via di rilevamento energetico che riduce la produzione di colesterolo e prosciuga i lipidi necessari a mantenere membrane neuronali e sinapsi robuste.
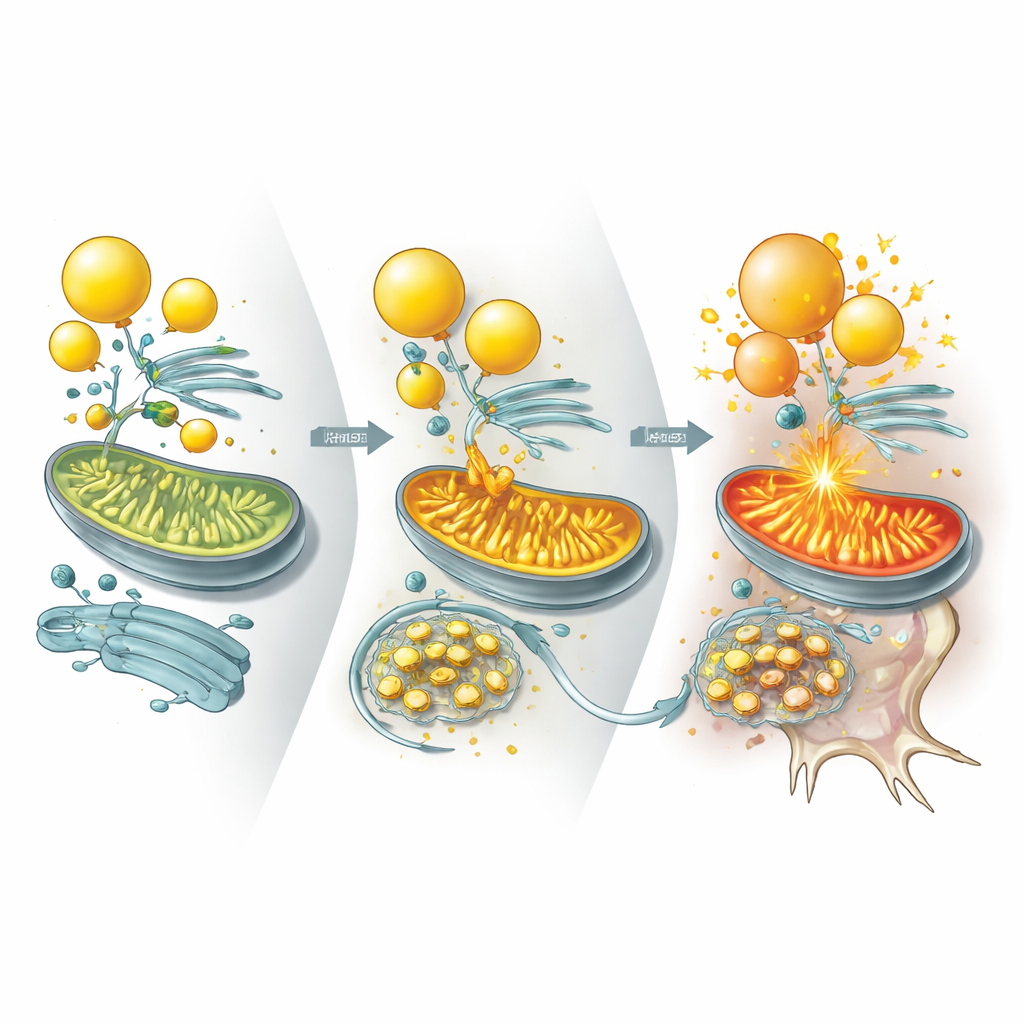
Prove da mini‑cervelli, topi e tessuti di pazienti
Lo stesso schema è emerso in più sistemi sperimentali. Gli organoidi cerebrali con UBQLN2 mutato svilupparono meno goccioline lipidiche e mostrarono maggiore morte neuronale sotto stress energetico, che poteva nuovamente essere attenuata con l’integrazione di colesterolo. In topi ingegnerizzati per esprimere una variante di UBQLN2 legata alla malattia, i neuroni accumularono ILVBL e ALDH3A2, persero goccioline lipidiche e degenerarono, portando a problemi motori e mnemonici. Importante, ridurre i livelli di ILVBL o ALDH3A2 in questi topi ripristinò le riserve lipidiche, migliorò la sopravvivenza neuronale e in parte recuperò la funzione motoria. Gli autori hanno anche esaminato tessuto del midollo spinale di pazienti con SLA sporadica caratterizzata da aggregati della proteina TDP‑43. In questi campioni, UBQLN2 risultava intrappolato nelle inclusioni di TDP‑43, ILVBL e ALDH3A2 si accumulavano, le goccioline lipidiche erano esaurite e i lipidi legati al colesterolo erano ridotti — rispecchiando i modelli sperimentali.
Trasformare un circolo vizioso in un bersaglio
Per i non specialisti, il messaggio chiave è che alcune forme di SLA e FTD possono riflettere non solo ammassi proteici tossici ma anche un lento collasso dell’economia energetica e delle membrane del neurone. UBQLN2 normalmente tiene gli enzimi che processano i lipidi sotto controllo in modo che la combustione dei grassi sostenga, anziché sabotare, la salute cellulare. Quando UBQLN2 è mutato o sequestrato dagli aggregati di TDP‑43, questi enzimi agiscono senza freni, le goccioline lipidiche si riducono, la produzione di colesterolo cala e le sinapsi si indeboliscono — un circolo vizioso che sfocia nella neurodegenerazione. Identificando questo asse UBQLN2–ILVBL/ALDH3A2 e dimostrando che sia la riduzione di questi enzimi sia il ripristino del colesterolo possono essere protettivi, lo studio apre nuove strade terapeutiche per rallentare o prevenire la perdita di neuroni in SLA, FTD e malattie correlate.
Citazione: Liu, Y., Huang, Z., Hsu, YW. et al. UBQLN2 links proteotoxicity with lipid metabolism in neurodegeneration. Nat Neurosci 29, 782–795 (2026). https://doi.org/10.1038/s41593-026-02226-y
Parole chiave: ALS, demenza frontotemporale, metabolismo dei lipidi, omeostasi proteica, neurodegenerazione