Clear Sky Science · pl
UBQLN2 łączy proteotoksyczność z metabolizmem lipidów w neurodegeneracji
Jak komórki nerwowe tracą paliwo
Dlaczego niektóre komórki nerwowe obumierają w chorobach takich jak stwardnienie zanikowe boczne (ALS) i otępienie czołowo‑skroniowe (FTD)? To badanie pokazuje, że pojedyncze białko pełniące rolę „policjanta ruchu” w komórce łączy dwa kluczowe zadania neuronów: usuwanie uszkodzonych białek oraz zarządzanie tłuszczami i cholesterolem. Gdy ten „policjant” zawodzi, komórki nerwowe szkodliwie spalają swoje zapasy tłuszczu, tracą istotne składniki budulcowe błon i ostatecznie obumierają. Zrozumienie tego ukrytego kryzysu energetycznego może wskazać nowe sposoby na dłuższe utrzymanie wrażliwych komórek mózgowych przy życiu.
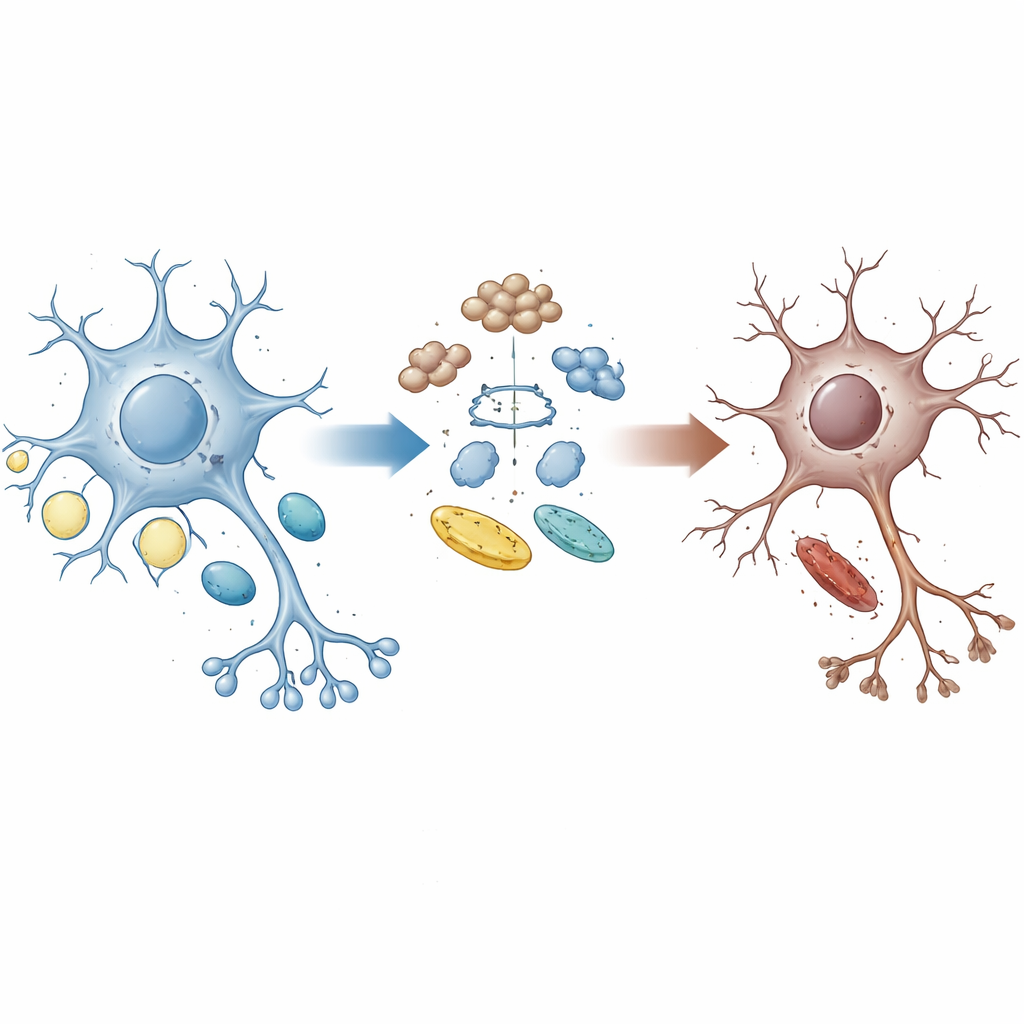
Zerwane ogniwo między porządkowaniem a energią
Zdrowe komórki mózgowe nieustannie recyklingują zużyte białka i dostosowują wykorzystanie cukrów oraz tłuszczów do swoich potrzeb energetycznych. Białko UBQLN2 działa jak kurier, kierując uszkodzone lub zbędne białka do komórkowego systemu utylizacji. Rodziny z określonymi mutacjami w UBQLN2 mogą rozwijać ALS i FTD, co sugeruje, że awarie tego systemu porządkowego mogą napędzać chorobę. Autorzy użyli ludzkich neuronów ruchowych pochodzących ze szczepionych komórek macierzystych z mutacjami UBQLN2 przypominającymi te u pacjentów, a także organoidów mózgu i modeli mysich, aby zbadać, jak to białko kształtuje równowagę między kontrolą jakości białek a metabolizmem energetycznym.
Gdy glukoza się kończy, spalanie tłuszczu idzie nie w tę stronę
Ponieważ mózgi w ALS i FTD często wykazują upośledzone wykorzystanie glukozy, zespół obciążył swoje neurony ruchowe poprzez usunięcie glukozy, zmuszając komórki do większego polegania na tłuszczach. Korzystając z połączenia proteomiki, lipidomiki i sekwencjonowania RNA, odkryli, że mutacje UBQLN2 spowalniają obrót tysięcy białek i silnie zmieniają ścieżki związane z metabolizmem. Pod wpływem stresu energetycznego zmutowane neurony zwiększały utlenianie długołańcuchowych kwasów tłuszczowych w mitochondriach i wykazywały wyraźny ubytek kropelek lipidowych — malutkich rezerwuarów tłuszczu — oraz lipidów bogatych w cholesterol. Pęcherzyki synaptyczne, małe woreczki uwalniające sygnały chemiczne między komórkami nerwowymi, stały się kruche, a przeżywalność neuronów spadła. Dodanie umiarkowanych ilości cholesterolu pomogło przywrócić integralność pęcherzyków i chronić zmutowane neurony oraz organoidy mózgu, podkreślając utratę cholesterolu jako kluczowy element problemu.
Dwa enzymy przechylają szalę
Idąc dalej, badacze zapytali, które konkretne białka UBQLN2 zwykle kontroluje, aby utrzymać metabolizm lipidów pod kontrolą. Zidentyfikowali dwa enzymy, ILVBL i ALDH3A2, które pomagają przekształcać określone cząsteczki tłuszczowe w formy gotowe do spalenia w mitochondriach. W zdrowych komórkach UBQLN2 wiąże te enzymy i kieruje je do degradacji, zwłaszcza w warunkach stresu, ograniczając, jak intensywnie tłuszcze są dostarczane do mitochondriów. W neuronach z mutacją UBQLN2 natomiast ILVBL i ALDH3A2 utrzymują się znacznie dłużej i kumulują się w miejscach kontaktu między kroplami lipidowymi a mitochondriami. To napędza nadmierne spalanie tłuszczu, aktywuje szlak sensingowy energetyczny, który tłumi produkcję cholesterolu, i odpompowuje lipidy niezbędne do utrzymania solidnych błon neuronalnych i synaps.
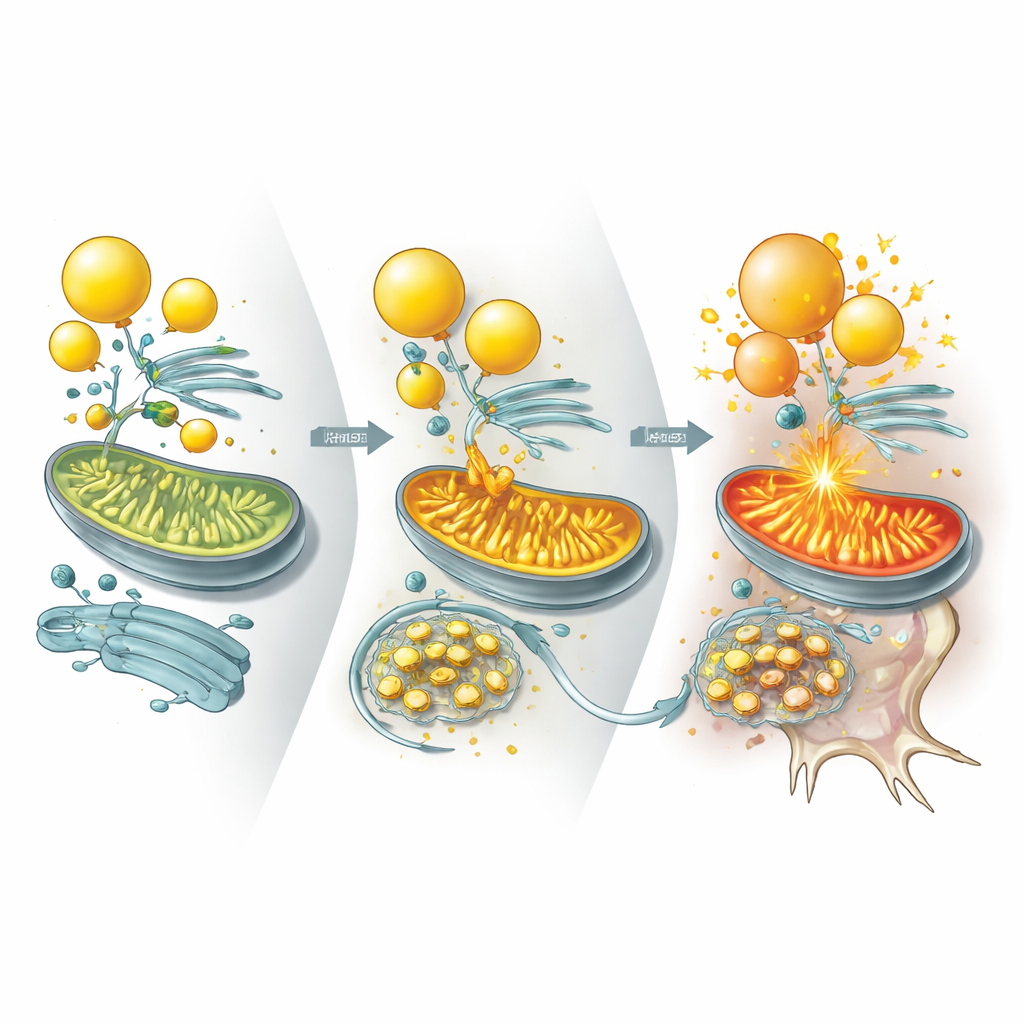
Dowody z mini‑mózgów, myszy i tkanek pacjentów
Ten sam wzorzec pojawił się w różnych systemach. Organoidy mózgowe z mutacją UBQLN2 rozwijały mniej kropelek lipidowych i wykazywały więcej śmierci neuronów pod wpływem stresu energetycznego, co ponownie można było złagodzić przez suplementację cholesterolu. U myszek zaprojektowanych tak, by wyrażać wariant UBQLN2 związany z chorobą, neurony gromadziły ILVBL i ALDH3A2, traciły krople lipidowe i degenerowały, prowadząc do problemów motorycznych i pamięciowych. Co ważne, obniżenie poziomów ILVBL lub ALDH3A2 u tych myszy przywracało zapasy lipidów, poprawiało przeżywalność neuronów i częściowo ratowało zdolności ruchowe. Autorzy przebadali także ludzkie tkanki rdzenia kręgowego od pacjentów ze sporadycznym ALS oznaczonym agregatami białka TDP‑43. W tych próbkach UBQLN2 było uwięzione w inkluzjach TDP‑43, ILVBL i ALDH3A2 się nagromadziły, krople lipidowe zostały wyczerpane, a lipidy związane z cholesterolem zmniejszone — odzwierciedlając modele eksperymentalne.
Przekształcanie błędnego koła w cel terapeutyczny
Dla osób niebędących specjalistami kluczowy przekaz jest taki, że niektóre postaci ALS i FTD mogą odzwierciedlać nie tylko toksyczne grudki białkowe, lecz także powolny upadek gospodarki paliwowej i błonowej neuronu. UBQLN2 zwykle trzyma enzymy przetwarzające lipidy krótko na smyczy, tak aby spalanie tłuszczu wspierało, a nie sabotowało zdrowie komórki. Gdy UBQLN2 jest zmutowane lub związane przez agregaty TDP‑43, te enzymy działają bez kontroli, krople lipidowe kurczą się, produkcja cholesterolu spada, a synapsy słabną — błędne koło kończące się neurodegeneracją. Wskazanie osi UBQLN2–ILVBL/ALDH3A2 oraz pokazanie, że zarówno osłabienie aktywności tych enzymów, jak i przywrócenie cholesterolu może być ochronne, otwiera nowe możliwości terapeutyczne spowalniające lub zapobiegające utracie komórek nerwowych w ALS, FTD i chorobach pokrewnych.
Cytowanie: Liu, Y., Huang, Z., Hsu, YW. et al. UBQLN2 links proteotoxicity with lipid metabolism in neurodegeneration. Nat Neurosci 29, 782–795 (2026). https://doi.org/10.1038/s41593-026-02226-y
Słowa kluczowe: ALS, otępienie czołowo‑skroniowe, metabolizm lipidów, homeostaza białek, neurodegeneracja