Clear Sky Science · fr
UBQLN2 relie la protéotoxicité au métabolisme des lipides dans la neurodégénérescence
Comment les neurones manquent de carburant
Pourquoi certains neurones meurent‑ils dans des maladies comme la sclérose latérale amyotrophique (SLA) et la démence frontotemporale (DFT) ? Cette étude montre qu’une seule protéine cellulaire, agissant comme un « agent de circulation », relie deux fonctions vitales de nos neurones : l’élimination des protéines endommagées et la gestion des graisses et du cholestérol. Lorsque cette protéine fait défaut, les neurones consument leurs réserves de lipides de façon délétère, perdent des composants membranaires essentiels et finissent par mourir. Comprendre cette crise énergétique cachée pourrait ouvrir de nouvelles voies pour préserver plus longtemps les cellules cérébrales vulnérables.
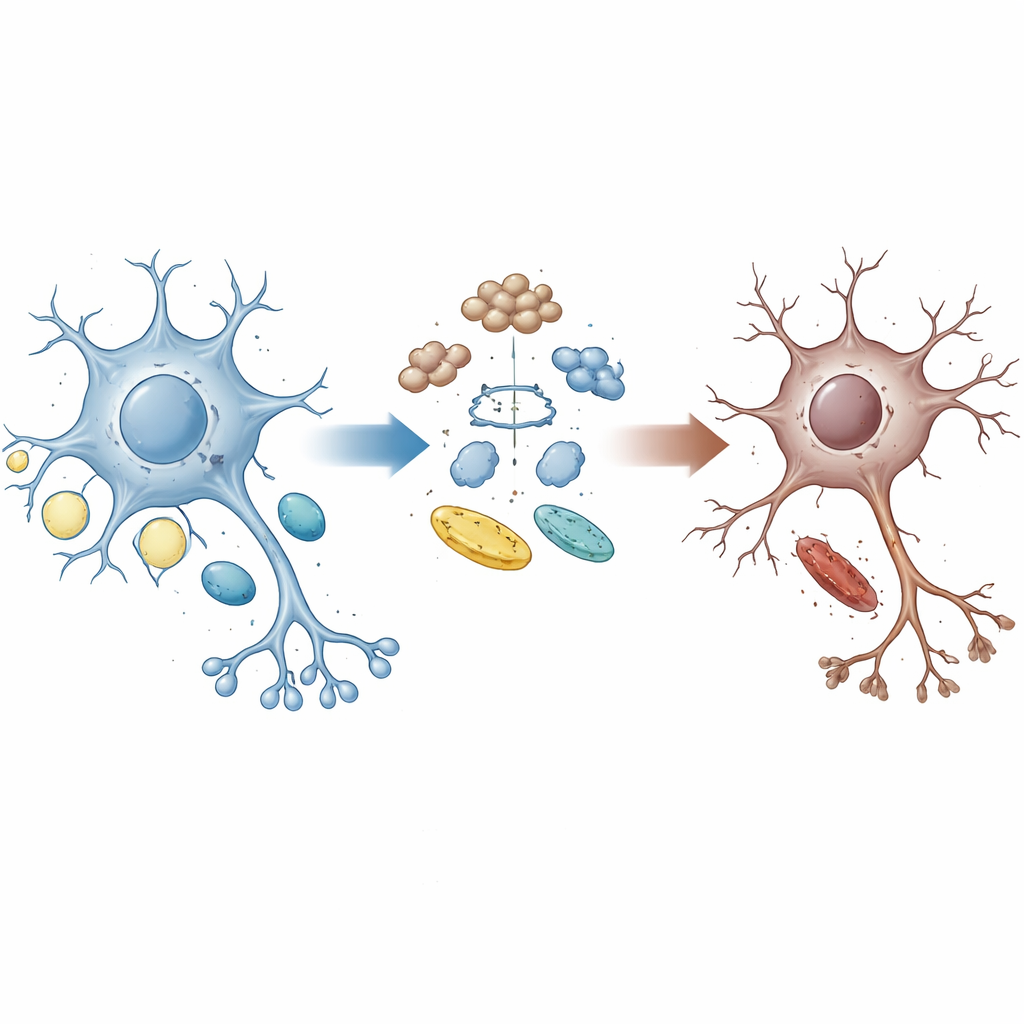
Un lien rompu entre le nettoyage et l’énergie
Les cellules cérébrales saines recyclent en permanence les protéines usées et ajustent l’utilisation des sucres et des lipides pour répondre à leurs besoins énergétiques. La protéine UBQLN2 joue le rôle de navette qui guide les protéines endommagées ou superflues vers les systèmes d’élimination de la cellule. Des familles porteuses de certaines mutations d’UBQLN2 développent la SLA et la DFT, ce qui suggère que des défaillances de ce système de nettoyage participent à la maladie. Les auteurs ont utilisé des motoneurones dérivés de cellules souches humaines portant des mutations UBQLN2 similaires à celles des patients, ainsi que des organoïdes cérébraux et des modèles murins, pour étudier comment cette protéine influence l’équilibre entre le contrôle de la qualité des protéines et le métabolisme énergétique.
Quand le glucose vient à manquer, la combustion des graisses déraille
Comme les cerveaux atteints de SLA et de DFT présentent souvent un mauvais usage du glucose, l’équipe a soumis ses motoneurones à un stress en éliminant le glucose, contraignant les cellules à dépendre davantage des lipides. Grâce à une combinaison de protéomique, lipidomique et séquençage ARN, ils ont constaté que les mutations d’UBQLN2 ralentissent le renouvellement de milliers de protéines et modifient fortement les voies liées au métabolisme. Sous stress énergétique, les neurones mutants ont accru l’oxydation des acides gras à longue chaîne dans les mitochondries et montré une perte marquée de gouttelettes lipidiques — petites réserves de graisse — et de lipides riches en cholestérol. Les vésicules synaptiques, ces petits sacs qui libèrent les signaux chimiques entre neurones, sont devenues fragiles, et la survie neuronale a diminué. Un apport modeste de cholestérol a permis de restaurer l’intégrité des vésicules et de protéger les neurones mutants et les organoïdes, soulignant la perte de cholestérol comme élément clé du problème.
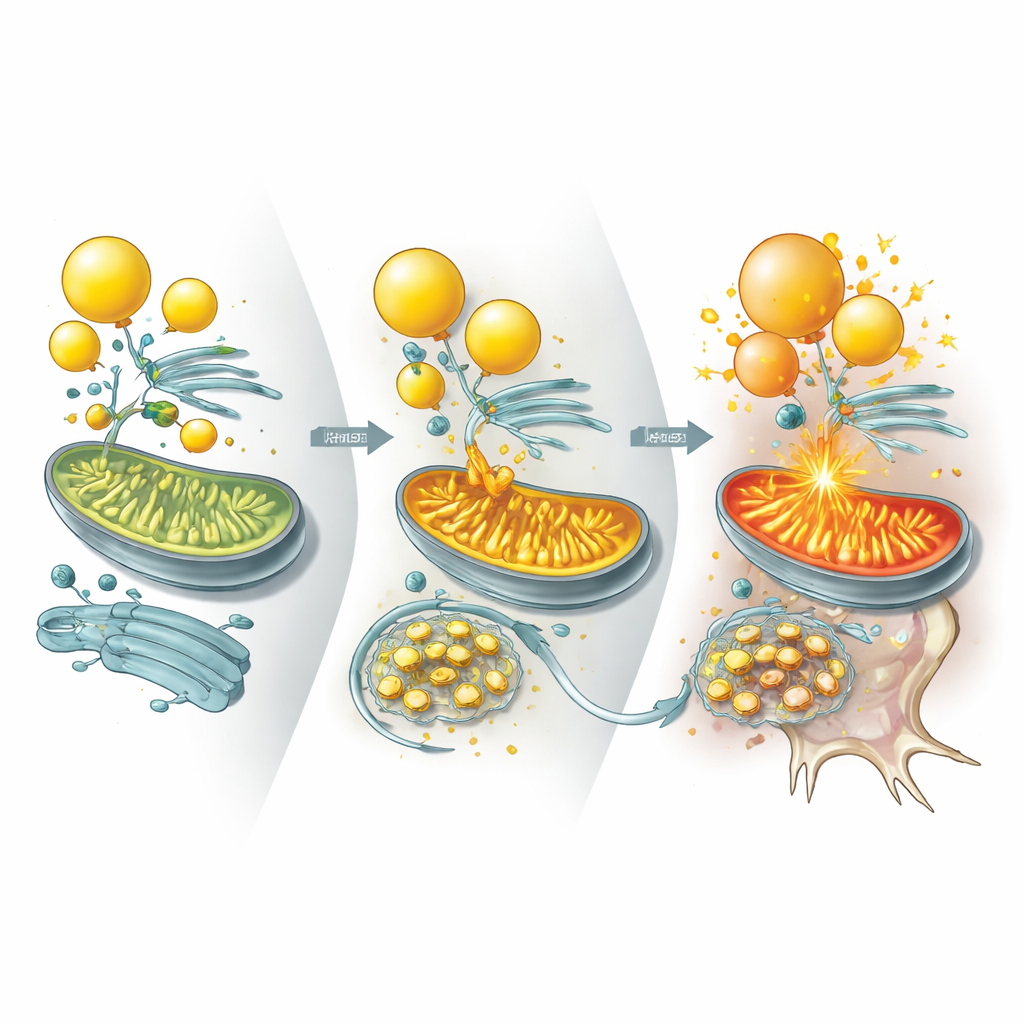
Deux enzymes font basculer l’équilibre
En creusant plus loin, les chercheurs ont cherché quelles protéines spécifiques UBQLN2 contrôle normalement pour réguler le métabolisme lipidique. Ils ont identifié deux enzymes, ILVBL et ALDH3A2, qui participent à la conversion de certains lipides en formes prêtes à être oxydées dans les mitochondries. Dans les cellules saines, UBQLN2 se lie à ces enzymes et les oriente vers la dégradation, en particulier lors du stress, limitant ainsi l’apport excessif de graisses aux mitochondries. Dans les neurones mutants pour UBQLN2, cependant, ILVBL et ALDH3A2 persistent beaucoup plus longtemps et s’accumulent aux points de contact entre les gouttelettes lipidiques et les mitochondries. Cela provoque une combustion excessive des lipides, active une voie de détection énergétique qui réduit la production de cholestérol, et épuise les lipides nécessaires au maintien de membranes neuronales et de synapses robustes.
Preuves tirées de mini‑cerveaux, de souris et de tissus de patients
Le même schéma est apparu dans plusieurs systèmes. Les organoïdes cérébraux mutants pour UBQLN2 ont développé moins de gouttelettes lipidiques et montré davantage de mort neuronale sous stress énergétique, une situation qui pouvait encore une fois être atténuée par une supplémentation en cholestérol. Chez des souris modifiées pour exprimer une variante d’UBQLN2 liée à la maladie, les neurones ont accumulé ILVBL et ALDH3A2, perdu leurs gouttelettes lipidiques et dégénéré, entraînant des troubles moteurs et mnésiques. Fait important, réduire les niveaux d’ILVBL ou d’ALDH3A2 chez ces souris a restauré les réserves lipidiques, amélioré la survie neuronale et partiellement sauvé la motricité. Les auteurs ont aussi examiné des tissus de moelle épinière humaine provenant de patients atteints de SLA sporadique marquée par des agrégats de la protéine TDP‑43. Dans ces échantillons, UBQLN2 était piégé dans les inclusions de TDP‑43, ILVBL et ALDH3A2 s’étaient accumulés, les gouttelettes lipidiques étaient appauvries et les lipides liés au cholestérol réduits — reflétant les modèles expérimentaux.
Transformer un cercle vicieux en cible thérapeutique
Pour un public non spécialiste, le message clé est que certaines formes de SLA et de DFT peuvent refléter non seulement des amas protéiques toxiques mais aussi un effondrement progressif de l’économie énergétique et membranaire du neurone. UBQLN2 maintient normalement les enzymes de traitement des lipides sous contrôle afin que la combustion des graisses soutienne, plutôt que sabote, la santé cellulaire. Quand UBQLN2 est muté ou séquestré par des agrégats de TDP‑43, ces enzymes deviennent incontrôlées, les gouttelettes lipidiques rétrécissent, la production de cholestérol chute et les synapses s’affaiblissent — un cercle vicieux qui aboutit à la neurodégénérescence. En identifiant cet axe UBQLN2–ILVBL/ALDH3A2 et en montrant que soit réduire l’activité de ces enzymes, soit restaurer le cholestérol peut être protecteur, l’étude ouvre de nouvelles pistes thérapeutiques pour ralentir ou prévenir la perte de neurones dans la SLA, la DFT et des maladies apparentées.
Citation: Liu, Y., Huang, Z., Hsu, YW. et al. UBQLN2 links proteotoxicity with lipid metabolism in neurodegeneration. Nat Neurosci 29, 782–795 (2026). https://doi.org/10.1038/s41593-026-02226-y
Mots-clés: SLA, démence frontotemporale, métabolisme des lipides, homéostasie des protéines, neurodégénérescence