Clear Sky Science · de
UBQLN2 verbindet Proteotoxizität mit Lipidstoffwechsel bei Neurodegeneration
Wie Nervenzellen die Energie ausgeht
Warum sterben in Erkrankungen wie amyotropher Lateralsklerose (ALS) und frontotemporaler Demenz (FTD) bestimmte Nervenzellen ab? Diese Studie zeigt, dass ein einzelnes zelluläres „Verkehrsdirigent“-Protein zwei lebenswichtige Aufgaben in unseren Neuronen verknüpft: das Beseitigen beschädigter Proteine und das Management von Fetten und Cholesterin. Wenn dieser Verkehrsdirigent versagt, verbrennen Nervenzellen ihre Fettspeicher auf schädliche Weise, verlieren wichtige Bausteine für Membranen und sterben letztlich. Das Verständnis dieser verborgenen Energiekrise könnte auf neue Wege hinweisen, verletzliche Gehirnzellen länger zu erhalten.
Eine gebrochene Verbindung zwischen Reinigung und Energie
Gesunde Gehirnzellen recyceln ständig verschlissene Proteine und passen ihre Nutzung von Zucker und Fetten an, um ihren Energiebedarf zu decken. Das Protein UBQLN2 fungiert als Shuttle, das beschädigte oder überflüssige Proteine zur zellulären Entsorgungsmaschinerie leitet. Familien mit bestimmten UBQLN2‑Mutationen können ALS und FTD entwickeln, was darauf hindeutet, dass Fehler in diesem Reinigungssystem zur Krankheit beitragen könnten. Die Autorinnen und Autoren nutzten motorische Neuronen aus menschlichen Stammzellen mit patientenähnlichen UBQLN2‑Mutationen sowie Hirnorganoide und Mausmodelle, um zu untersuchen, wie dieses Protein das Gleichgewicht zwischen Proteinqualitätskontrolle und Energiestoffwechsel prägt.
Wenn Glukose knapp wird, gerät Fettverbrennung außer Kontrolle
Da im Gehirn von ALS‑ und FTD‑Patienten oft eine schlechte Glukosenutzung beobachtet wird, setzten die Forschenden ihre motorischen Neuronen unter Stress, indem sie Glukose entfernten und die Zellen zwangen, stärker auf Fette zurückzugreifen. Mit einer Kombination aus Proteomik, Lipidomik und RNA‑Sequenzierung fanden sie heraus, dass UBQLN2‑Mutationen den Umsatz von Tausenden Proteinen verlangsamen und Stoffwechselwege stark verändern. Unter Energiestress erhöhten die mutierten Neuronen die mitochondriale Oxidation von langkettigen Fettsäuren und zeigten einen deutlichen Verlust an Lipidtröpfchen—kleinen Fettspeichern—und cholesterinreichen Lipiden. Synaptische Vesikel, die kleinen Säckchen, die chemische Signale zwischen Nervenzellen freisetzen, wurden fragil, und das Überleben der Neuronen sank. Die Zugabe moderater Cholesterinmengen half, die Vesikelintegrität wiederherzustellen und die mutierten Neuronen und Hirnorganoide zu schützen, was den Cholesterinverlust als Schlüsselaspekt des Problems hervorhebt.
Zwei Enzyme kippen das Gleichgewicht
Bei genauerer Betrachtung fragten die Forschenden, welche spezifischen Proteine UBQLN2 normalerweise kontrolliert, um den Lipidstoffwechsel in Schach zu halten. Sie identifizierten zwei Enzyme, ILVBL und ALDH3A2, die bestimmte Fettmoleküle in Formen umwandeln, die in den Mitochondrien verbrannt werden können. In gesunden Zellen bindet UBQLN2 diese Enzyme und lenkt sie, besonders unter Stress, zur Degradation, wodurch begrenzt wird, wie aggressiv Fette in die Mitochondrien eingespeist werden. In UBQLN2‑mutierten Neuronen hingegen bleiben ILVBL und ALDH3A2 deutlich länger erhalten und sammeln sich an Kontaktstellen zwischen Lipidtröpfchen und Mitochondrien an. Das treibt übermäßige Fettverbrennung an, aktiviert einen Energiesensor‑Signalweg, der die Cholesterinproduktion drosselt, und raubt den Lipiden die nötige Substanz, um robuste neuronale Membranen und Synapsen zu erhalten.
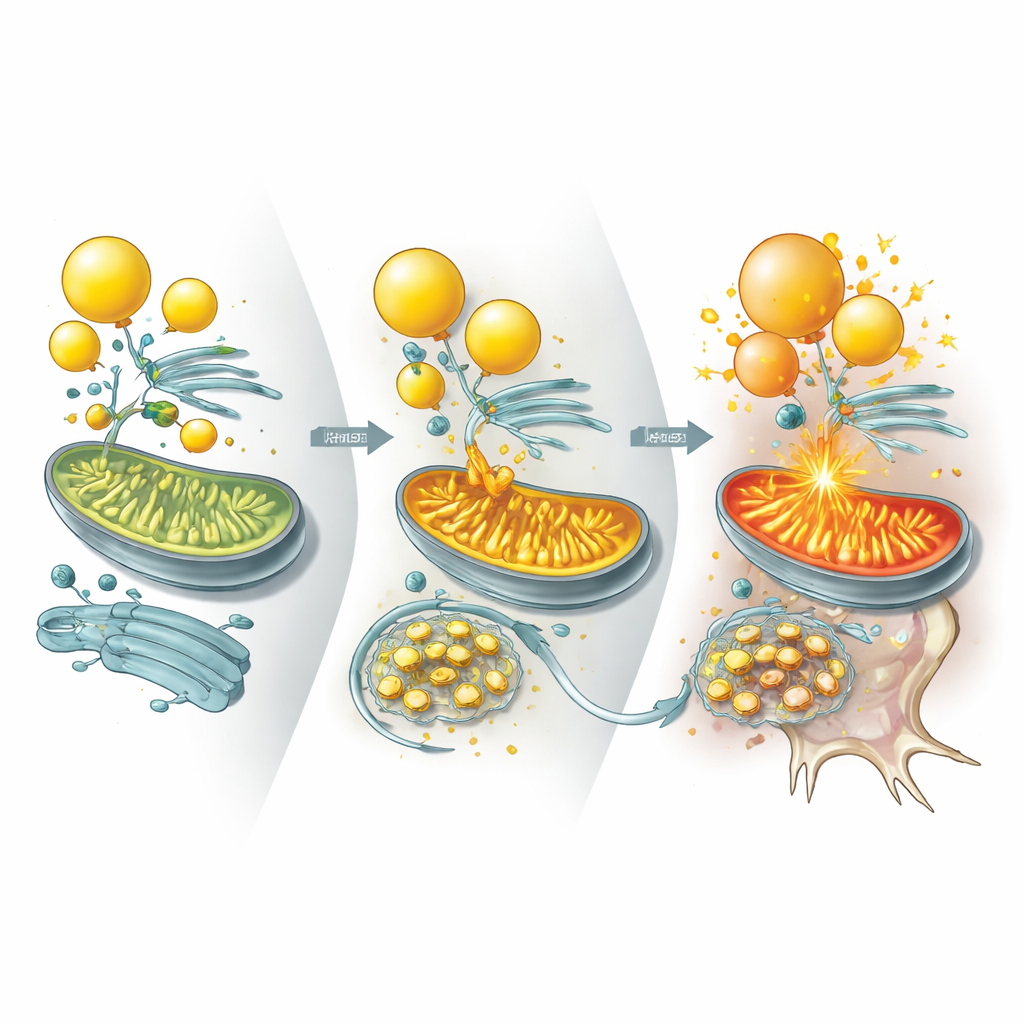
Hinweise aus Mini‑Gehirnen, Mäusen und Patientenproben
Dasselbe Muster zeigte sich in mehreren Systemen. UBQLN2‑mutante Hirnorganoide entwickelten weniger Lipidtröpfchen und zeigten unter Energiestress mehr neuronales Absterben, was erneut durch Cholesterinzufuhr gemildert werden konnte. In Mäusen, die so verändert wurden, dass sie eine krankheitsassoziierte UBQLN2‑Variante exprimieren, sammelten Neuronen ILVBL und ALDH3A2 an, verloren Lipidtröpfchen und degenerierten, was zu motorischen und Gedächtnisstörungen führte. Wichtig ist, dass das Senken von ILVBL‑ oder ALDH3A2‑Spiegeln in diesen Mäusen die Lipidspeicher wiederherstellte, das Überleben der Neuronen verbesserte und die Beweglichkeit teilweise rettete. Die Autorinnen und Autoren untersuchten außerdem menschliches Rückenmarkgewebe von Patientinnen und Patienten mit sporadischer ALS, gekennzeichnet durch TDP‑43‑Proteinaggregate. In diesen Proben war UBQLN2 in TDP‑43‑Inklusionen gefangen, ILVBL und ALDH3A2 häuften sich an, Lipidtröpfchen waren erschöpft und cholesterinbezogene Lipide reduziert—ein Spiegelbild der experimentellen Modelle.
Aus einem Teufelskreis ein Ziel machen
Für Nicht‑Spezialisten ist die Kernbotschaft: Einige Formen von ALS und FTD spiegeln nicht nur toxische Protein‑Ablagerungen wider, sondern auch einen schleichenden Zusammenbruch der Energie‑ und Membranökonomie der Neuronen. UBQLN2 hält normalerweise Lipid‑verarbeitende Enzyme kurz, sodass Fettverbrennung die Zellgesundheit unterstützt, anstatt sie zu sabotieren. Wenn UBQLN2 mutiert ist oder durch TDP‑43‑Aggregate sequestriert wird, laufen diese Enzyme unkontrolliert, Lipidtröpfchen schrumpfen, die Cholesterinproduktion sinkt und Synapsen schwächen sich—ein Teufelskreis, der in Neurodegeneration endet. Indem die Studie diese UBQLN2–ILVBL/ALDH3A2‑Achse identifiziert und zeigt, dass sowohl das Herunterregeln dieser Enzyme als auch die Wiederherstellung von Cholesterin schützend wirken kann, eröffnet sie neue therapeutische Ansätze, um den Verlust von Nervenzellen bei ALS, FTD und verwandten Erkrankungen zu verlangsamen oder zu verhindern.
Zitation: Liu, Y., Huang, Z., Hsu, YW. et al. UBQLN2 links proteotoxicity with lipid metabolism in neurodegeneration. Nat Neurosci 29, 782–795 (2026). https://doi.org/10.1038/s41593-026-02226-y
Schlüsselwörter: ALS, frontotemporale Demenz, Lipidstoffwechsel, Protein‑Homöostase, Neurodegeneration