Clear Sky Science · en
UBQLN2 links proteotoxicity with lipid metabolism in neurodegeneration
How Nerve Cells Run Out of Fuel
Why do some nerve cells die in disorders like amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)? This study shows that a single cellular "traffic cop" protein helps link two vital jobs inside our neurons: clearing damaged proteins and managing fats and cholesterol. When this traffic cop goes wrong, nerve cells burn their fat stores in a harmful way, lose key membrane building blocks, and ultimately die. Understanding this hidden energy crisis could point to new ways to keep vulnerable brain cells alive longer.
A Broken Link Between Cleanup and Energy
Healthy brain cells constantly recycle worn‑out proteins and adjust how they use sugars and fats to meet their energy needs. The protein UBQLN2 acts as a shuttle that guides damaged or unneeded proteins to the cell’s disposal machinery. Families with certain UBQLN2 mutations can develop ALS and FTD, suggesting that failures in this cleanup system might help drive disease. The authors used human stem‑cell–derived motor neurons carrying patient‑like UBQLN2 mutations, along with brain organoids and mouse models, to see how this protein shapes the balance between protein quality control and energy metabolism.
When Glucose Runs Low, Fat Burning Goes Off the Rails
Because ALS and FTD brains often show poor glucose use, the team stressed their motor neurons by removing glucose, forcing cells to rely more on fats. Using a combination of proteomics, lipidomics and RNA sequencing, they found that UBQLN2 mutations slow the turnover of thousands of proteins and strongly alter pathways related to metabolism. Under energy stress, mutant neurons ramped up long‑chain fatty‑acid oxidation in mitochondria and showed a marked loss of lipid droplets—tiny fat reservoirs—and cholesterol‑rich lipids. Synaptic vesicles, the small sacs that release chemical signals between nerve cells, became fragile, and neuron survival dropped. Adding back modest amounts of cholesterol helped restore vesicle integrity and protect mutant neurons and brain organoids, highlighting cholesterol loss as a key part of the problem.
Two Enzymes Tip the Balance
Diving deeper, the researchers asked which specific proteins UBQLN2 normally controls to keep lipid metabolism in check. They identified two enzymes, ILVBL and ALDH3A2, that help convert certain fatty molecules into forms ready to be burned in mitochondria. In healthy cells, UBQLN2 binds these enzymes and steers them toward degradation, especially during stress, limiting how aggressively fats are fed into mitochondria. In UBQLN2‑mutant neurons, however, ILVBL and ALDH3A2 linger much longer and accumulate at contact sites between lipid droplets and mitochondria. This drives excessive fat burning, activates an energy‑sensing pathway that dampens cholesterol production, and drains the lipids needed to maintain robust neuronal membranes and synapses.
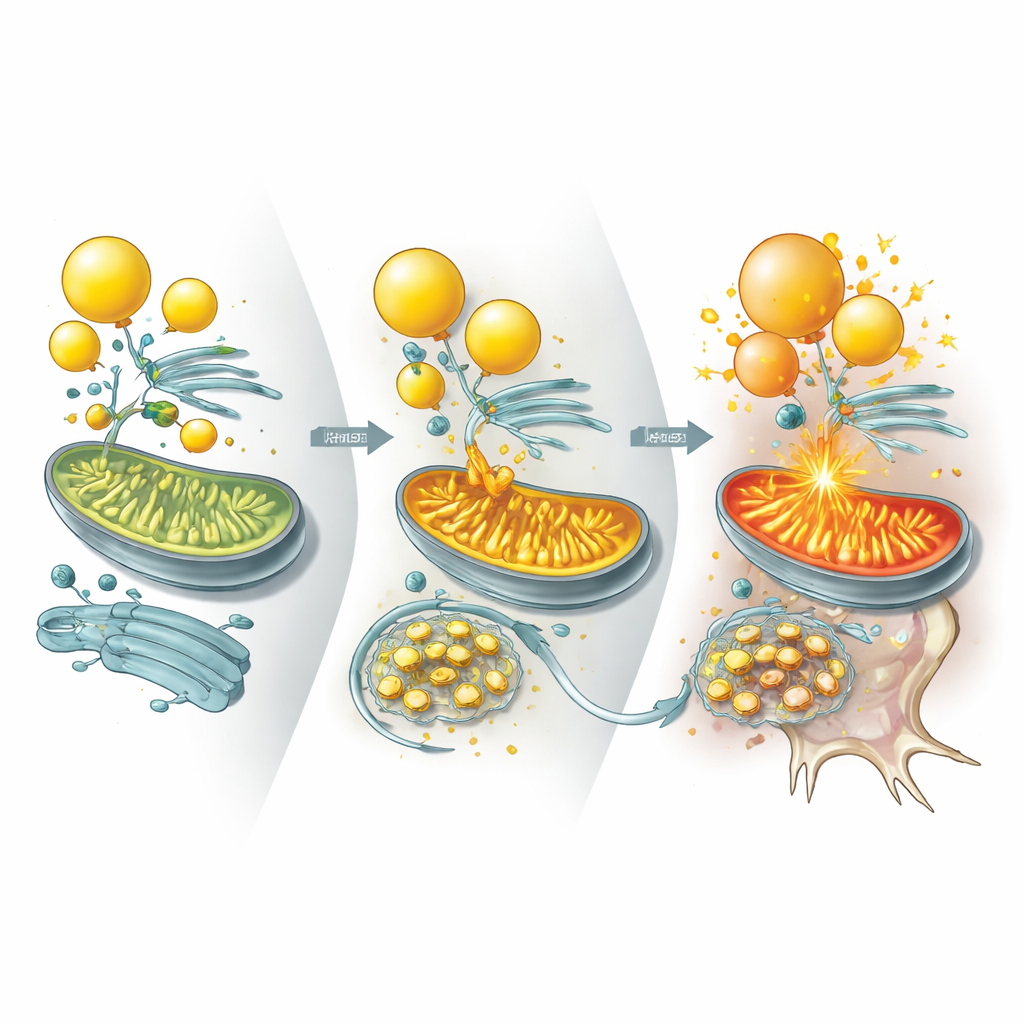
Evidence from Mini‑Brains, Mice, and Patient Tissues
The same pattern emerged across multiple systems. UBQLN2‑mutant brain organoids developed fewer lipid droplets and showed more neuronal death under energy stress, which could again be eased by cholesterol supplementation. In mice engineered to express a disease‑linked UBQLN2 variant, neurons accumulated ILVBL and ALDH3A2, lost lipid droplets, and degenerated, leading to motor and memory problems. Importantly, lowering ILVBL or ALDH3A2 levels in these mice restored lipid stores, improved neuron survival, and partially rescued movement. The authors also examined human spinal cord tissue from patients with sporadic ALS marked by TDP‑43 protein aggregates. In these samples, UBQLN2 was trapped in TDP‑43 inclusions, ILVBL and ALDH3A2 piled up, lipid droplets were depleted, and cholesterol‑related lipids were reduced—mirroring the experimental models.
Turning a Vicious Cycle into a Target
For non‑specialists, the key message is that some forms of ALS and FTD may reflect not just toxic protein clumps but also a slow collapse of the neuron’s fuel and membrane economy. UBQLN2 normally keeps lipid‑processing enzymes on a short leash so that fat burning supports, rather than sabotages, cell health. When UBQLN2 is mutated or sequestered by TDP‑43 aggregates, these enzymes run unchecked, lipid droplets shrink, cholesterol production drops, and synapses weaken—a vicious cycle that ends in neurodegeneration. By pinpointing this UBQLN2–ILVBL/ALDH3A2 axis, and showing that either dialing down these enzymes or restoring cholesterol can be protective, the study opens up new therapeutic angles for slowing or preventing nerve cell loss in ALS, FTD and related diseases.
Citation: Liu, Y., Huang, Z., Hsu, YW. et al. UBQLN2 links proteotoxicity with lipid metabolism in neurodegeneration. Nat Neurosci 29, 782–795 (2026). https://doi.org/10.1038/s41593-026-02226-y
Keywords: ALS, frontotemporal dementia, lipid metabolism, protein homeostasis, neurodegeneration