Clear Sky Science · es
UBQLN2 vincula la proteotoxicidad con el metabolismo de los lípidos en la neurodegeneración
Cómo las neuronas se quedan sin combustible
¿Por qué mueren algunas neuronas en trastornos como la esclerosis lateral amiotrófica (ELA) y la demencia frontotemporal (DFT)? Este estudio muestra que una sola proteína celular, a modo de "agente de tráfico", conecta dos funciones vitales dentro de nuestras neuronas: la eliminación de proteínas dañadas y la gestión de grasas y colesterol. Cuando este agente falla, las neuronas consumen sus reservas de grasa de forma perjudicial, pierden componentes esenciales de las membranas y, en última instancia, mueren. Entender esta crisis energética oculta podría señalar nuevas vías para mantener con vida por más tiempo a las células cerebrales vulnerables.
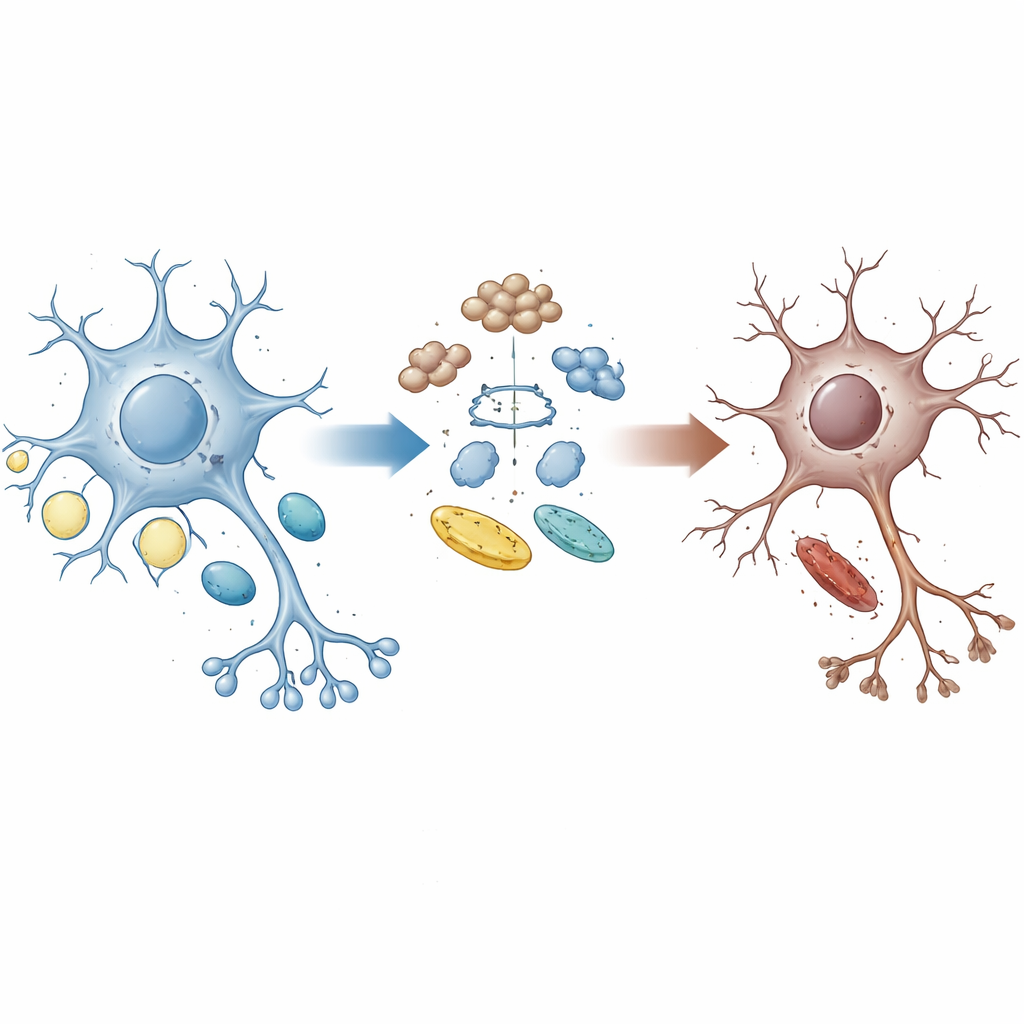
Un vínculo roto entre la limpieza y la energía
Las células cerebrales sanas reciclan constantemente proteínas desgastadas y ajustan cómo usan azúcares y grasas para cubrir sus necesidades energéticas. La proteína UBQLN2 actúa como un transportador que guía las proteínas dañadas o innecesarias hacia el sistema de eliminación de la célula. Familias con ciertas mutaciones en UBQLN2 pueden desarrollar ELA y DFT, lo que sugiere que fallos en este sistema de limpieza podrían contribuir a la enfermedad. Los autores emplearon neuronas motoras derivadas de células madre humanas portadoras de mutaciones en UBQLN2 semejantes a las de pacientes, junto con organoides cerebrales y modelos de ratón, para estudiar cómo esta proteína modula el equilibrio entre el control de calidad proteico y el metabolismo energético.
Cuando la glucosa escasea, la quema de grasa se descontrola
Como en el cerebro de pacientes con ELA y DFT suele haber un uso deficiente de glucosa, el equipo estresó sus neuronas motoras retirando la glucosa, obligando a las células a depender más de las grasas. Mediante una combinación de proteómica, lipidómica y secuenciación de ARN, encontraron que las mutaciones en UBQLN2 ralentizan la renovación de miles de proteínas y alteran de forma marcada las vías relacionadas con el metabolismo. Bajo estrés energético, las neuronas mutantes aumentaron la oxidación de ácidos grasos de cadena larga en las mitocondrias y mostraron una pérdida notable de gotas lipídicas —pequeños depósitos de grasa— y de lípidos ricos en colesterol. Las vesículas sinápticas, los pequeños sacos que liberan señales químicas entre las neuronas, se volvieron frágiles y la supervivencia neuronal disminuyó. Reponer cantidades modestas de colesterol ayudó a restaurar la integridad de las vesículas y protegió a las neuronas mutantes y a los organoides cerebrales, lo que subraya la pérdida de colesterol como una pieza central del problema.
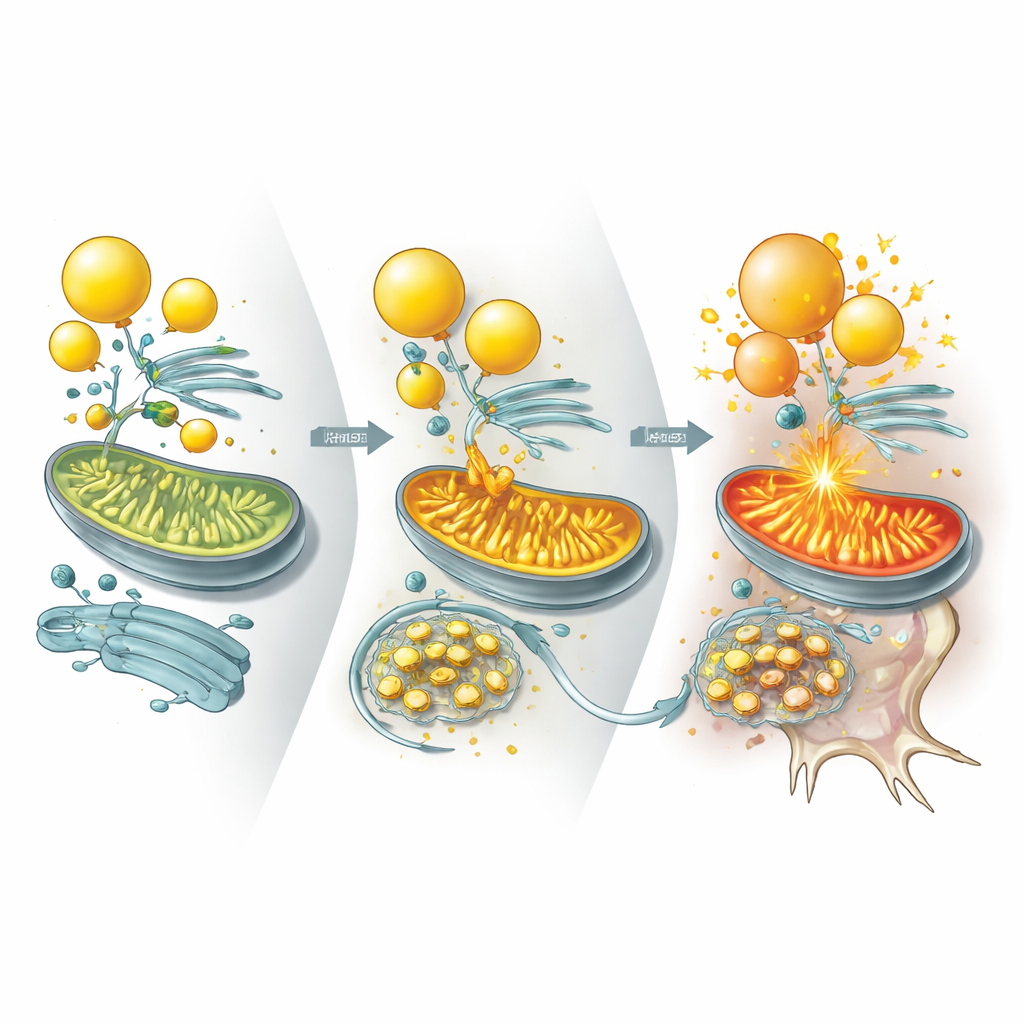
Dos enzimas inclinan la balanza
Profundizando, los investigadores preguntaron qué proteínas concretas controla normalmente UBQLN2 para mantener el metabolismo lipídico bajo control. Identificaron dos enzimas, ILVBL y ALDH3A2, que ayudan a convertir ciertos lípidos en formas aptas para ser quemadas en las mitocondrias. En células sanas, UBQLN2 se une a estas enzimas y las dirige hacia la degradación, especialmente durante el estrés, limitando cuán agresivamente las grasas se canalizan a las mitocondrias. En neuronas con mutación en UBQLN2, sin embargo, ILVBL y ALDH3A2 persisten mucho más tiempo y se acumulan en los sitios de contacto entre las gotas lipídicas y las mitocondrias. Esto impulsa una quema excesiva de grasas, activa una vía sensora de energía que reduce la producción de colesterol y agota los lípidos necesarios para mantener membranas y sinapsis neuronales robustas.
Pruebas en mini‑cerebros, ratones y tejidos de pacientes
El mismo patrón emergió en múltiples sistemas. Los organoides cerebrales con mutación en UBQLN2 desarrollaron menos gotas lipídicas y mostraron más muerte neuronal bajo estrés energético, lo que pudo aliviarse nuevamente con suplementación de colesterol. En ratones diseñados para expresar una variante de UBQLN2 asociada a la enfermedad, las neuronas acumularon ILVBL y ALDH3A2, perdieron gotas lipídicas y degeneraron, conduciendo a problemas motores y de memoria. De forma importante, reducir los niveles de ILVBL o ALDH3A2 en estos ratones restauró las reservas lipídicas, mejoró la supervivencia neuronal y rescató parcialmente la movilidad. Los autores también examinaron tejido de médula espinal humana de pacientes con ELA esporádica marcada por agregados de proteína TDP‑43. En estas muestras, UBQLN2 estaba atrapada en inclusiones de TDP‑43, ILVBL y ALDH3A2 se acumulaban, las gotas lipídicas estaban agotadas y los lípidos relacionados con el colesterol se reducían —reflejando los modelos experimentales.
Convertir un ciclo vicioso en un objetivo
Para el público general, el mensaje clave es que algunas formas de ELA y DFT pueden reflejar no solo acúmulos proteicos tóxicos, sino también un colapso gradual de la economía energética y de membranas de la neurona. UBQLN2 normalmente mantiene a las enzimas que procesan lípidos bajo control para que la quema de grasas apoye, en lugar de sabotee, la salud celular. Cuando UBQLN2 está mutada o secuestrada por agregados de TDP‑43, estas enzimas actúan sin freno, las gotas lipídicas se encogen, la producción de colesterol cae y las sinapsis se debilitan —un ciclo vicioso que termina en neurodegeneración. Al identificar este eje UBQLN2–ILVBL/ALDH3A2 y mostrar que tanto reducir estas enzimas como restaurar el colesterol puede ser protector, el estudio abre nuevas vías terapéuticas para ralentizar o prevenir la pérdida de neuronas en ELA, DFT y enfermedades relacionadas.
Cita: Liu, Y., Huang, Z., Hsu, YW. et al. UBQLN2 links proteotoxicity with lipid metabolism in neurodegeneration. Nat Neurosci 29, 782–795 (2026). https://doi.org/10.1038/s41593-026-02226-y
Palabras clave: ELA, demencia frontotemporal, metabolismo de lípidos, homeostasis de proteínas, neurodegeneración