Clear Sky Science · sv
Skalbara atomära grundmodeller via beskärning av meddelandeöverföring och grafuppdelning
Varför snabbare atomfilmer spelar roll
Många av de tekniker vi bryr oss om, från bättre batterier till renare katalysatorer, beror på vad atomer gör över långa tider och i stora partikelantal. Datorsimuleringar kan skapa ”filmer” av dessa atomer i rörelse, men de mest exakta metoderna är så långsamma att de ofta stannar vid mycket små system och mycket korta tidsskalor. Denna artikel presenterar ett sätt att behålla den höga noggrannheten hos moderna artificiella intelligensmodeller för atomer samtidigt som kostnaden minskas så mycket att simuleringar med miljontals atomer och nanosekundlängder blir praktiska på dagens grafikprocessorer. 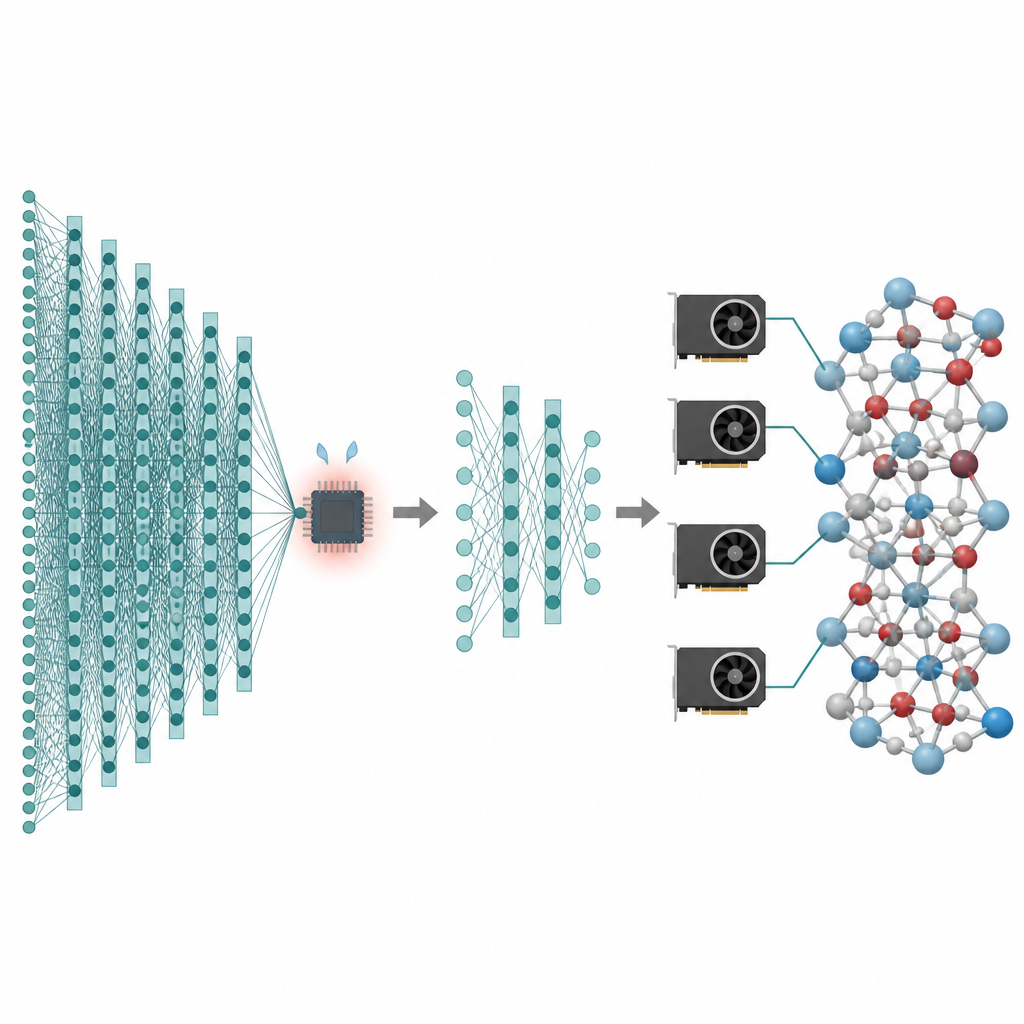
Från kvantkopplad noggrannhet till praktiska gränser
I ett ytterlighetsfall beskriver kvantbaserade metoder atomära interaktioner med stor detaljrikedom men är långt för långsamma för stora, realistiska system. För att överbrygga detta gap har forskare vänt sig till maskininlärningsmodeller som lär sig hur atomer påverkar varandra från kvantdata och sedan ersätter dyra beräkningar med snabba förutsägelser. Ett nyligt genombrott inom detta fält är framväxten av ”atomistiska grundmodeller”, stora neurala nätverk tränade på omfattande och varierade samlingar av atomstrukturer. Dessa modeller kan finjusteras med bara en liten mängd ny data och ändå matcha eller överträffa många specialbyggda modeller i noggrannhet, vilket gör dem attraktiva som allmänna verktyg för materialvetenskap.
När större modeller blir en börda
Styrkan hos dessa grundmodeller är också deras svaghet. För att täcka många grundämnen och bindningsmiljöer förlitar de sig på djupa ”message-passing”-nätverk som upprepade gånger utbyter information mellan närliggande atomer. Varje extra lager hjälper atomer att uppfatta ett större omgivande område, men det ökar också modellens storlek och minnesanvändning. Som ett resultat körs dessa modeller mycket långsammare än enklare maskininlärningspotentialer och kan inte lätt hantera strukturer mycket utöver tiotusentals atomer. För forskare som behöver studera sällsynta händelser, långsam diffusion eller komplexa fasövergångar blir denna prestandagren sätt kraftfulla modeller till nischverktyg. Författarna strävar efter att behålla fördelarna hos grundmodellerna samtidigt som onödiga kostnader skärs bort.
Beskärning av neurala nät utan att förlora förmåga
Det första steget i det föreslagna arbetsflödet kallas beskärning av meddelandeöverföringsdjupet. Teamet analyserade hur mycket varje lager i flera populära atomistiska grundmodeller faktiskt förändrar atomernas interna beskrivning. De observerade en effekt av ”överutjämning”: efter att de tidiga lagren har gjort det mesta av arbetet bidrar senare lager bara med små, repetitiva justeringar. Genom att utnyttja detta behöll de helt enkelt de första lagren och tog bort resten, för att sedan finjustera den förkortade modellen på uppgiftsspecifika data. Tester på litiumfosfatglas, blandade kisel–silika-system och metallpartiklar på oxidsupporter visar att även modeller med bara ett eller två kvarvarande lager kan nå nästan samma noggrannhet som de fulla nätverken, samtidigt som de blir mycket lättare och snabbare. 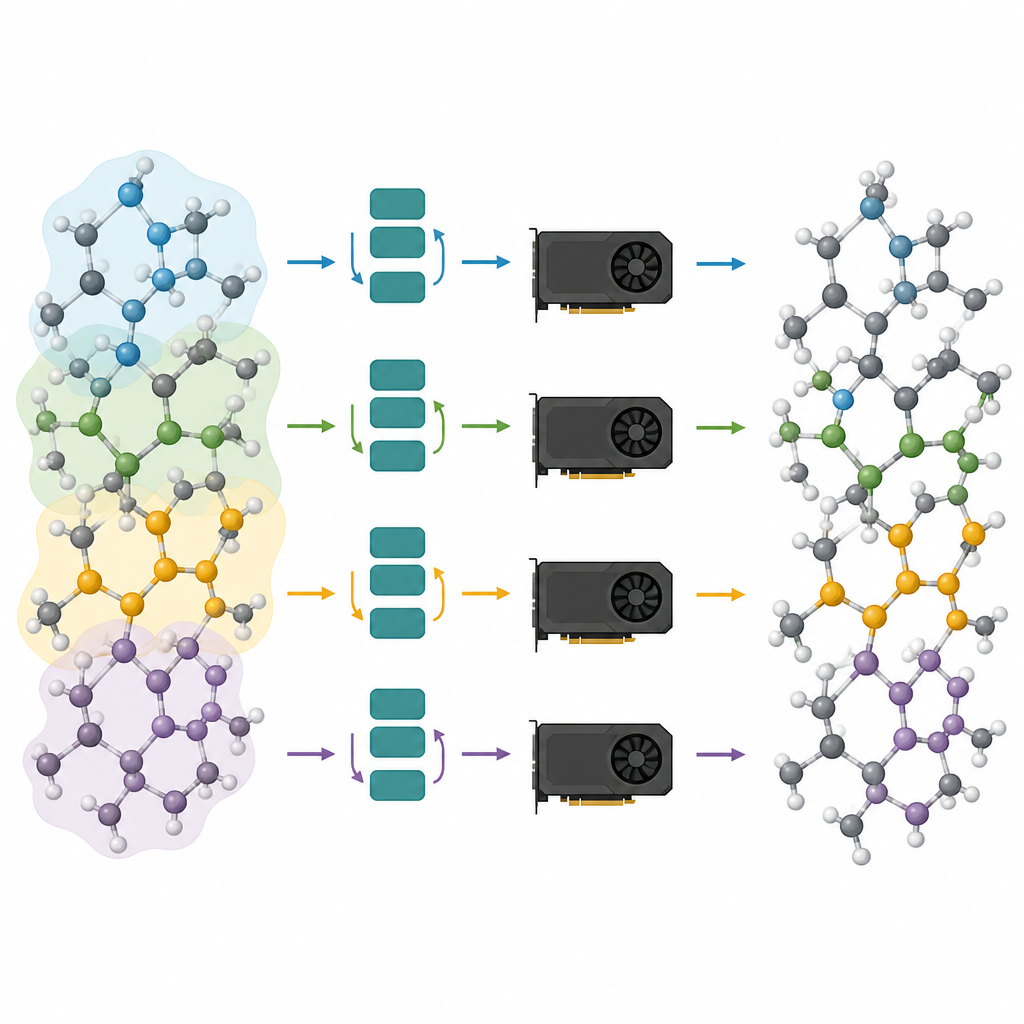
Att dela stora atomära system över många chip
Beskurna modeller är bara halva lösningen; den andra halvan är att köra dem effektivt på moderna grafikprocessorer. Författarna ser ett atomärt system som en graf, där atomer är noder och bindningar eller närliggande grannar är länkar. De delar upp denna graf i överlappande delar, var och en med alla grannar som behövs för ett valt antal meddelandeöverföringssteg, och tilldelar dessa delgrafer till olika grafikprocessorer. Eftersom modellen nu är grund blir det nödvändiga grannskapet litet och de extra atomerna i varje del förblir hanterbara. Avgörande är att varje processor kan köra sin del oberoende, utan behov av kommunikation under den tunga beräkningen, och resultaten kan sedan sammansättas. Denna typ av ”halo”-partitionering låter samma metod fungera oavsett om det finns ett enda grafikkort eller flera som arbetar parallellt.
Vad de nya verktygen möjliggör
Genom att kombinera beskärning med grafuppdelning uppnår författarna stora vinster i hastighet och skala. De visar att simuleringar med miljontals atomer och till och med system med mer än fem miljoner atomer är genomförbara på standardhårdvara, samtidigt som de bevarar de tillförlitliga krafter och strukturer som grundmodeller erbjuder. I praktiska fallstudier återger de beskurna modellerna bildandet av amorft litiumfosfat, nanoskalig separation i kisel–silika-glas och den gradvisa inneslutningen av en platina-nanopartikel av en defektfylld oxidyta. För icke-specialister är budskapet tydligt: detta arbete förvandlar tidigare långsamma, minneskrävande atomära AI:er till effektiva motorer som kan driva realistiska materialsimuleringar och öppnar dörren för rutinmässiga virtuella experiment på komplexa enheter och material.
Citering: Kong, L., Shim, J., Hu, G. et al. Scalable foundation interatomic potentials via message-passing pruning and graph partitioning. npj Comput Mater 12, 180 (2026). https://doi.org/10.1038/s41524-026-02001-4
Nyckelord: atomistiska grundmodeller, molekylär dynamik, grafneuronätverk, GPU-acceleration, materialsimulering