Clear Sky Science · de
Skalierbare atomare Grundpotenziale durch Message-Passing-Pruning und Graphpartitionierung
Warum schnellere atomare Filme wichtig sind
Viele Technologien, die uns am Herzen liegen – von besseren Batterien bis zu saubereren Katalysatoren – hängen davon ab, was Atome über lange Zeiträume und in sehr großen Teilchensammlungen tun. Computersimulationen können solche Atombewegungen als „Filme“ erzeugen, doch die genauesten Methoden sind so langsam, dass sie oft bei winzigen Systemen und sehr kurzen Zeiten stecken bleiben. Diese Arbeit stellt einen Weg vor, die hohe Genauigkeit moderner KI‑Modelle für Atome zu bewahren und gleichzeitig die Kosten so weit zu senken, dass Simulationen mit Millionen von Atomen über Nanosekunden auf heutigen Grafikprozessoren praktikabel werden. 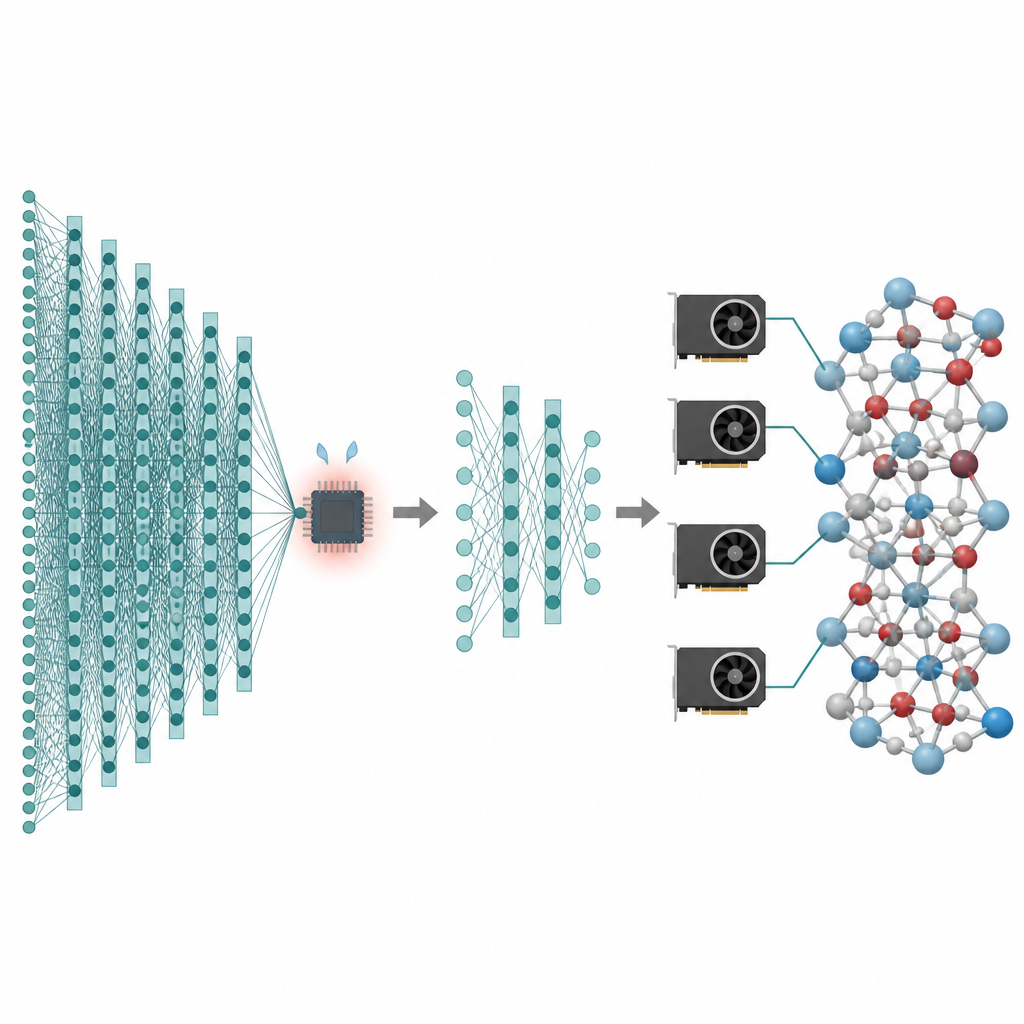
Von quantenmechanischer Genauigkeit zu praktischen Grenzen
Am einen Ende beschreiben quantenbasierte Methoden Atominteraktionen sehr detailliert, sind aber für große, realistische Systeme viel zu langsam. Um diese Lücke zu schließen, setzen Forscher auf maschinelle Lernmodelle, die aus quantenmechanischen Daten lernen, wie Atome einander beeinflussen, und teure Rechnungen durch schnelle Vorhersagen ersetzen. Ein jüngster Fortschritt in diesem Bereich sind „atomistische Grundmodelle“: große neuronale Netze, die auf umfangreichen und vielfältigen Sammlungen atomarer Strukturen trainiert wurden. Diese Modelle lassen sich mit nur wenigen neuen Daten feinabstimmen und erreichen oft die Genauigkeit spezialisierter Modelle oder übertreffen sie, was sie als universelle Werkzeuge für die Materialwissenschaft attraktiv macht.
Wenn größere Modelle zur Last werden
Die Stärke dieser Grundmodelle ist zugleich ihre Schwäche. Um viele Elemente und Bindungsumgebungen abzudecken, nutzen sie tiefe Message‑Passing‑Netze, die wiederholt Informationen zwischen benachbarten Atomen austauschen. Jede zusätzliche Schicht hilft Atomen, eine weitere Umgebung zu erfassen, vergrößert aber auch Modellgröße und Speicherbedarf. Dadurch laufen diese Modelle deutlich langsamer als einfachere ML‑Potentiale und können kaum Strukturen jenseits einiger Zehntausend Atome bewältigen. Für Forschende, die seltene Ereignisse, langsame Diffusionen oder komplexe Phasenübergänge untersuchen müssen, wird diese Performanzgrenze zu einem Ausschlusskriterium. Die Autor:innen verfolgen das Ziel, die Vorteile der Grundmodelle zu erhalten, gleichzeitig aber überflüssige Kosten zu entfernen.
Das neuronale Netz stutzen, ohne die Leistungsfähigkeit zu verlieren
Der erste Schritt im vorgeschlagenen Workflow heißt Message‑Passing‑Depth‑Pruning. Das Team analysierte, wie stark jede Schicht in mehreren populären atomistischen Grundmodellen die interne Beschreibung der Atome tatsächlich verändert. Sie beobachteten einen „Over‑Smoothing“-Effekt: Nachdem die frühen Schichten den Großteil der Arbeit erledigt haben, bewirken spätere Schichten nur noch winzige, wiederholte Anpassungen. Daraus folgend behalten sie einfach die ersten wenigen Schichten und entfernen den Rest, um das verkürzte Modell anschließend auf aufgabenspezifischen Daten feinzujustieren. Tests an Lithiumphosphatglas, gemischten Silizium‑Silica‑Systemen und Metallpartikeln auf Oxidträgern zeigen, dass Modelle mit nur einer oder zwei verbleibenden Schichten nahezu die gleiche Genauigkeit wie die vollständigen Netze erreichen können, gleichzeitig aber deutlich leichter und schneller sind. 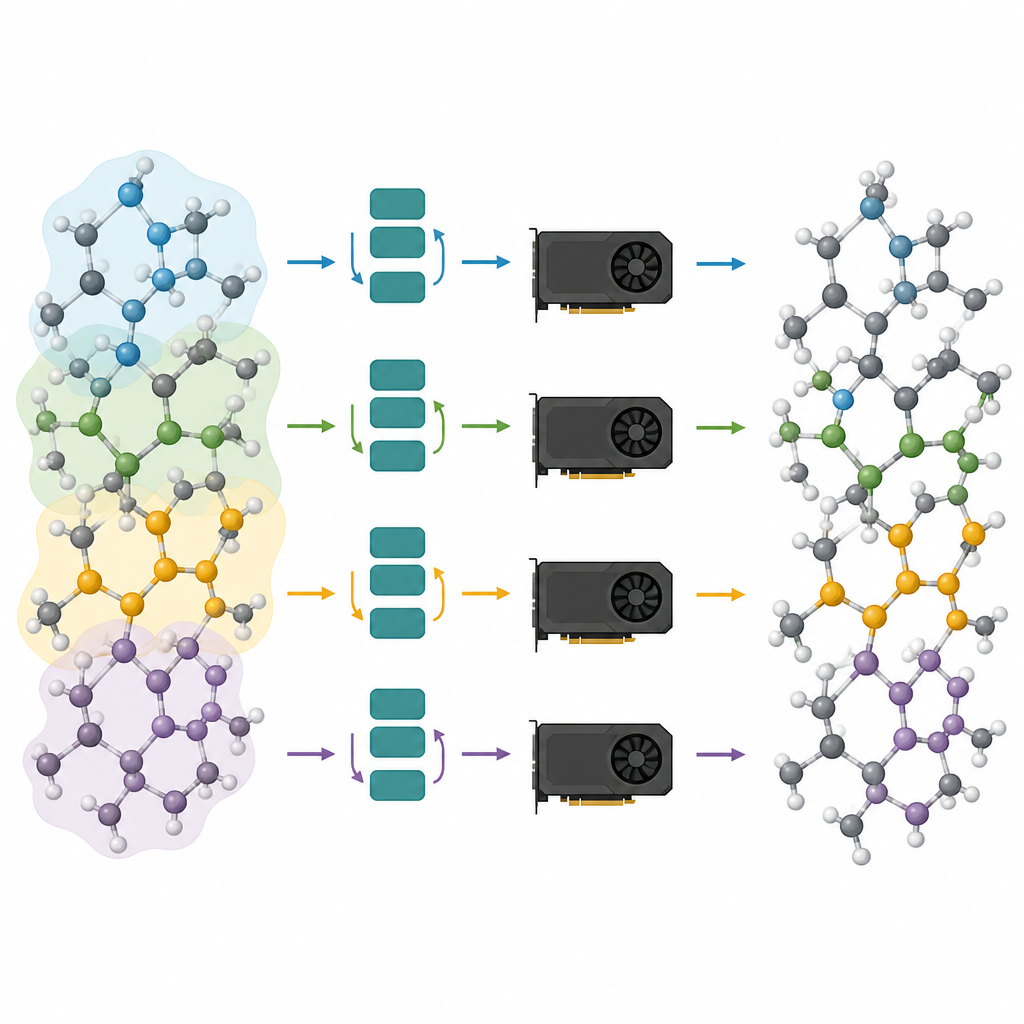
Große atomare Systeme über viele Chips verteilen
Beschnittene Modelle sind nur die halbe Lösung; die andere Hälfte besteht darin, sie effizient auf modernen Grafikprozessoren auszuführen. Die Autor:innen betrachten ein atomares System als Graph, bei dem Atome Knoten und Bindungen oder nahe Nachbarn Kanten sind. Sie partitionieren diesen Graphen in sich überlappende Teile, von denen jeder alle Nachbarn enthält, die für eine gewählte Anzahl von Message‑Passing‑Schritten nötig sind, und ordnen diese Teilgraphen verschiedenen GPUs zu. Da das Modell jetzt flach ist, ist die benötigte Nachbarschaft klein, und die zusätzlichen Atome in jedem Teilstück bleiben überschaubar. Entscheidend ist, dass jede GPU ihr Teilstück unabhängig berechnen kann, ohne während der rechenintensiven Phase kommunizieren zu müssen; die Ergebnisse werden anschließend zusammengefügt. Diese Halo‑artige Partitionierung erlaubt es, dieselbe Methode sowohl auf einer einzelnen Grafikkarte als auch auf mehreren parallel arbeitenden Karten anzuwenden.
Was die neuen Werkzeuge ermöglichen
Kombiniert man Pruning mit Graphpartitionierung, erzielen die Autor:innen große Gewinne bezüglich Geschwindigkeit und Skalierbarkeit. Sie zeigen, dass Simulationen mit einer Million Atomen und sogar Systeme mit mehr als fünf Millionen Atomen auf Standardhardware möglich sind, während die vertrauenswürdigen Kräfte und Strukturen, die Grundmodelle liefern, erhalten bleiben. In praxisnahen Fallstudien reproduzieren die beschnittenen Modelle die Bildung amorphen Lithiumphosphats, nanoskalige Trennung in Silizium‑Silica‑Gläsern und die schrittweise Einkapselung eines Platin‑Nanopartikels durch eine defekte Oxidoberfläche. Für Nicht‑Spezialist:innen ist die Botschaft klar: Diese Arbeit verwandelt zuvor langsame, speicherhungrige atomare KI‑Modelle in schlanke Motoren, die realistische Materialsimulationen antreiben können und den Weg für routinemäßige virtuelle Experimente an komplexen Bauteilen und Materialien öffnet.
Zitation: Kong, L., Shim, J., Hu, G. et al. Scalable foundation interatomic potentials via message-passing pruning and graph partitioning. npj Comput Mater 12, 180 (2026). https://doi.org/10.1038/s41524-026-02001-4
Schlüsselwörter: atomistische Grundmodelle, Molekulardynamik, Graphneuronale Netze, GPU‑Beschleunigung, Materialsimulation