Clear Sky Science · fr
Potentiels interatomiques fondamentaux évolutifs via élagage du message-passing et partitionnement de graphe
Pourquoi des films atomiques plus rapides comptent
Beaucoup des technologies qui nous intéressent, des batteries améliorées aux catalyseurs plus propres, dépendent du comportement des atomes sur de longues durées et à travers d’énormes nombres de particules. Les simulations informatiques peuvent créer des « films » montrant ces atomes en mouvement, mais les méthodes les plus précises sont si lentes qu’elles butent souvent sur des systèmes microscopiques et des durées très courtes. Cet article présente une façon de conserver la haute précision des modèles d’intelligence artificielle modernes pour les atomes tout en réduisant leur coût au point que des simulations de millions d’atomes sur des nanosecondes deviennent pratiques sur les processeurs graphiques d’aujourd’hui. 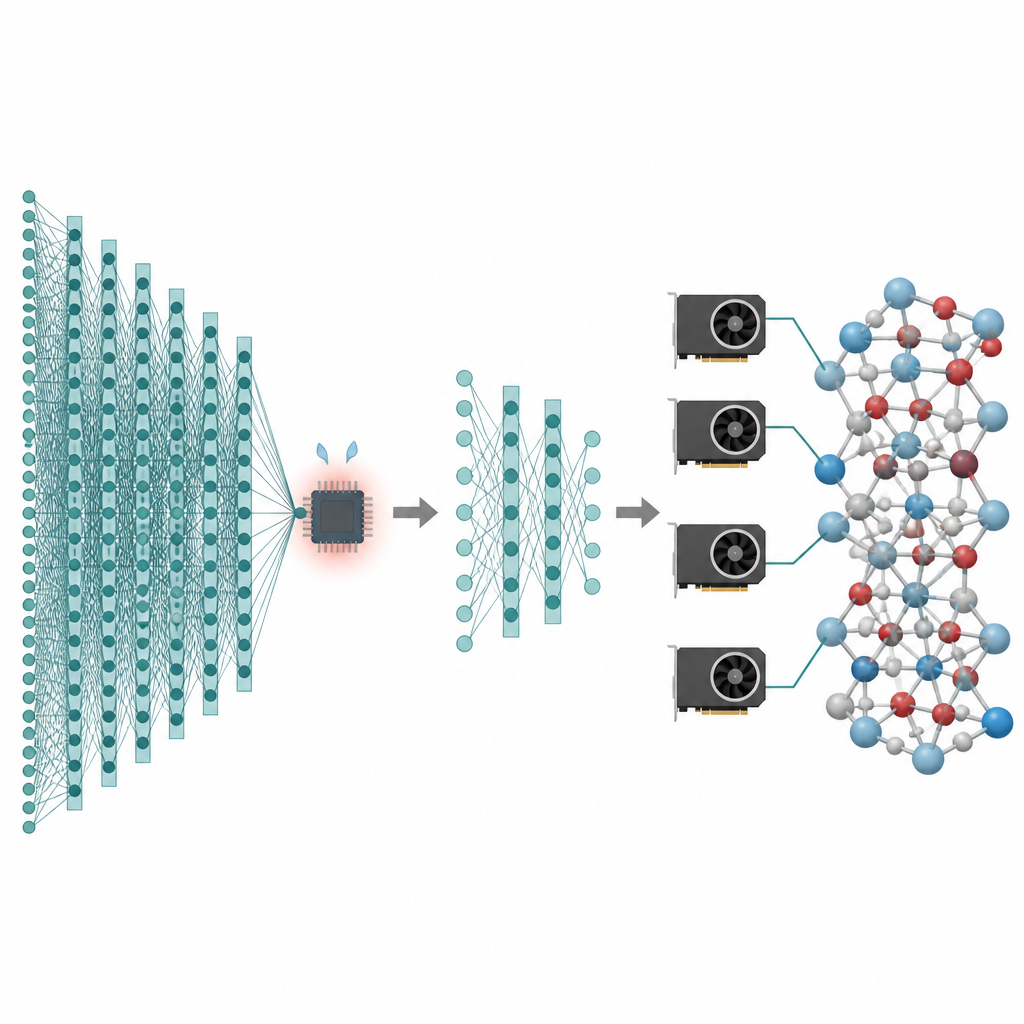
De la précision quantique aux limites pratiques
À une extrémité, les méthodes basées sur la mécanique quantique décrivent les interactions atomiques avec un grand niveau de détail mais sont beaucoup trop lentes pour des systèmes réalistes et étendus. Pour combler cet écart, les chercheurs se sont tournés vers des modèles d’apprentissage automatique qui apprennent, à partir de données quantiques, comment les atomes se poussent et se tirent, puis remplacent des calculs coûteux par des prédictions rapides. Une avancée récente dans ce domaine est l’émergence des « modèles fondamentaux atomistiques », de grands réseaux neuronaux entraînés sur d’immenses et variées collections de structures atomiques. Ces modèles peuvent être affinés avec une petite quantité de nouvelles données et égaler ou surpasser beaucoup de modèles conçus sur mesure en précision, ce qui les rend attractifs comme outils généraux pour la science des matériaux.
Quand les modèles plus grands deviennent un fardeau
La force de ces modèles fondamentaux est aussi leur faiblesse. Pour couvrir de nombreux éléments et environnements de liaison, ils reposent sur des réseaux profonds de « message-passing » qui échangent à plusieurs reprises de l’information entre atomes voisins. Chaque couche supplémentaire permet aux atomes de percevoir une région plus large autour d’eux, mais augmente aussi la taille et l’utilisation mémoire du modèle. En conséquence, ces modèles s’exécutent beaucoup plus lentement que des potentiels d’apprentissage automatique plus simples et ne peuvent pas gérer facilement des structures bien au-delà de quelques dizaines de milliers d’atomes. Pour les scientifiques qui doivent étudier des événements rares, une diffusion lente ou des changements de phase complexes, ce mur de performances transforme des modèles puissants en outils de niche. Les auteurs cherchent à conserver les bénéfices des modèles fondamentaux tout en supprimant les coûts inutiles.
Élaguer le réseau neuronal sans perdre ses compétences
La première étape du flux de travail proposé s’appelle l’élagage de la profondeur du message-passing. L’équipe a analysé dans quelle mesure chaque couche de plusieurs modèles fondamentaux atomistiques populaires modifie réellement la description interne des atomes. Ils ont observé un effet d’« over-smoothing » : après que les premières couches ont fait l’essentiel du travail, les couches ultérieures n’apportent que de minuscules ajustements répétitifs. En exploitant cela, ils conservent simplement les premières couches et suppriment le reste, puis affinent le modèle raccourci sur des données spécifiques à la tâche. Des tests sur du verre phosphate de lithium, des systèmes silicium–silice mixtes et des particules métalliques sur supports oxydes montrent que même des modèles ne conservant qu’une ou deux couches peuvent atteindre presque la même précision que les réseaux complets, tout en étant beaucoup plus légers et plus rapides. 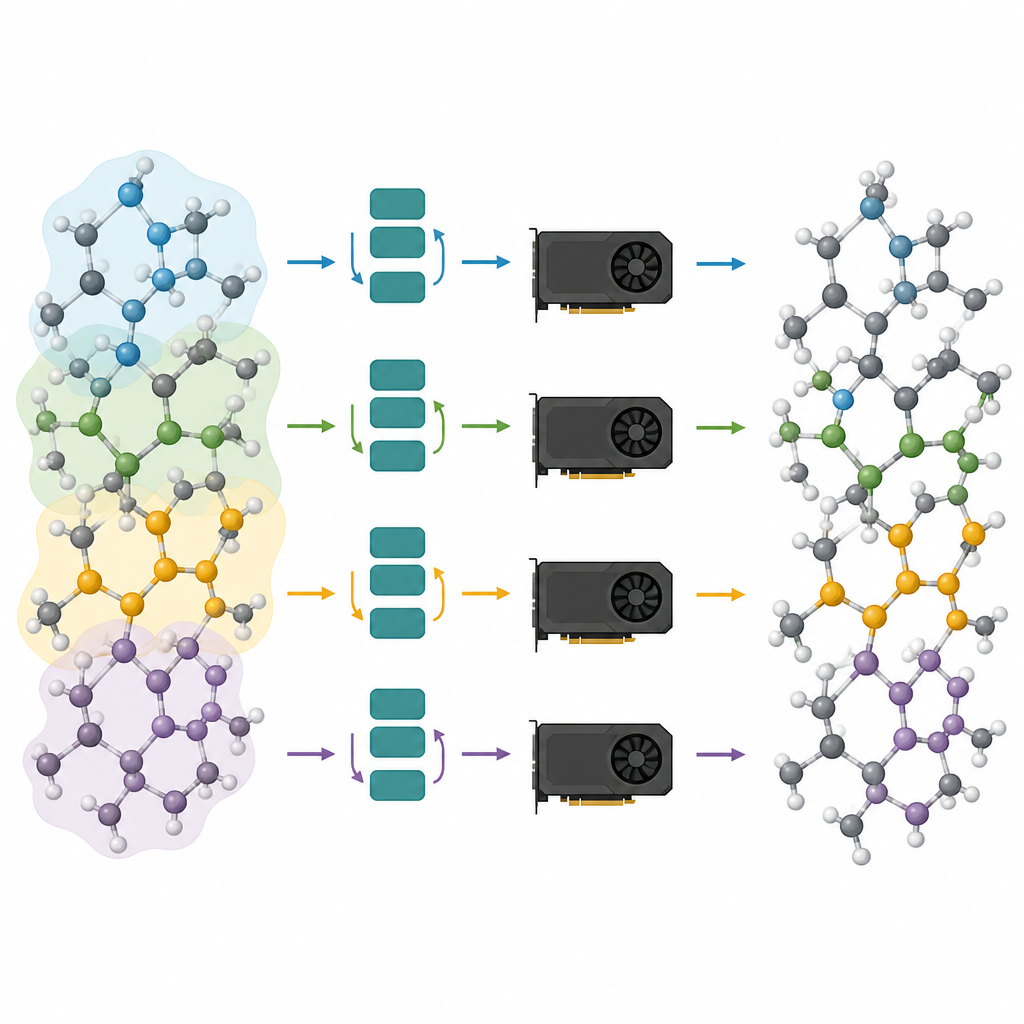
Répartir de grands systèmes atomiques sur plusieurs puces
Les modèles élagués ne constituent que la moitié de la solution ; l’autre moitié consiste à les exécuter efficacement sur les processeurs graphiques modernes. Les auteurs considèrent un système atomique comme un graphe, où les atomes sont des nœuds et les liaisons ou voisins rapprochés sont des liens. Ils partitionnent ce graphe en morceaux qui se chevauchent, chacun contenant tous les voisins nécessaires pour un nombre choisi d’étapes de message-passing, et assignent ces sous-graphes à différents processeurs graphiques. Parce que le modèle est désormais peu profond, le voisinage requis est réduit et les atomes supplémentaires dans chaque morceau restent gérables. Surtout, chaque processeur peut traiter son morceau de façon indépendante, sans besoin de communiquer pendant le calcul intensif, et les résultats sont ensuite recollés. Ce partitionnement de type « halo » permet à la même méthode de fonctionner qu’il y ait une seule carte graphique ou plusieurs en parallèle.
Ce que permettent les nouveaux outils
En combinant élagage et partitionnement de graphe, les auteurs obtiennent d’importants gains en vitesse et en échelle. Ils démontrent que des simulations à l’échelle du million d’atomes et même des systèmes de plus de cinq millions d’atomes sont réalisables sur du matériel standard, tout en maintenant les forces et structures fiables que fournissent les modèles fondamentaux. Dans des études de cas pratiques, les modèles élagués reproduisent la formation du phosphate de lithium amorphe, la séparation à l’échelle nanométrique dans des verres silicium–silice, et l’encapsulation progressive d’une nanoparticule de platine par une surface d’oxyde défectueuse. Pour les non-spécialistes, le message est clair : ce travail transforme des IA atomiques auparavant lentes et gourmandes en mémoire en moteurs sveltes capables d’alimenter des simulations réalistes de matériaux, ouvrant la voie à des expériences virtuelles routinières sur des dispositifs et matériaux complexes.
Citation: Kong, L., Shim, J., Hu, G. et al. Scalable foundation interatomic potentials via message-passing pruning and graph partitioning. npj Comput Mater 12, 180 (2026). https://doi.org/10.1038/s41524-026-02001-4
Mots-clés: modèles fondamentaux atomistiques, dynamique moléculaire, réseaux de neurones sur graphe, accélération GPU, simulation de matériaux