Clear Sky Science · en
Scalable foundation interatomic potentials via message-passing pruning and graph partitioning
Why faster atomic movies matter
Many of the technologies we care about, from better batteries to cleaner catalysts, depend on what atoms are doing over long times and across huge numbers of particles. Computer simulations can create “movies” of these atoms in motion, but the most accurate methods are so slow that they often stall at tiny systems and very short times. This paper presents a way to keep the high accuracy of modern artificial intelligence models for atoms while cutting their cost so much that million-atom, nanosecond-long simulations become practical on today’s graphics processors. 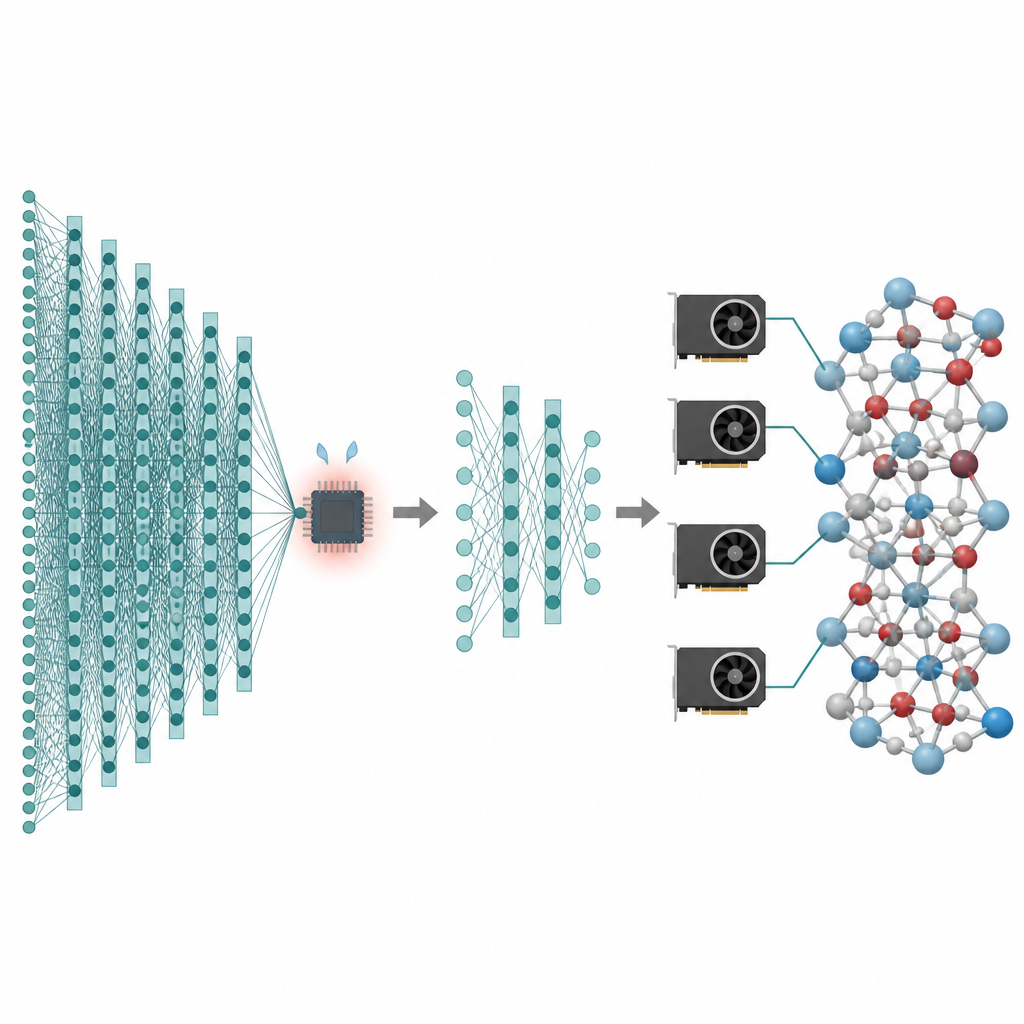
From quantum accuracy to practical limits
At one extreme, quantum-based methods describe atomic interactions with great detail but are far too slow for large, realistic systems. To bridge this gap, researchers have turned to machine learning models that learn how atoms push and pull on each other from quantum data, then replace expensive calculations with fast predictions. A recent leap in this field is the rise of “atomistic foundation models,” large neural networks trained on vast and varied collections of atomic structures. These models can be fine-tuned with only a small amount of new data and still match or beat many custom-built models in accuracy, making them attractive as general tools for materials science.
When bigger models become a burden
The strength of these foundation models is also their weakness. To cover many elements and bonding environments, they rely on deep “message-passing” networks that repeatedly exchange information between neighboring atoms. Each added layer helps atoms sense a wider region around them, but it also inflates the model’s size and memory use. As a result, these models run much slower than simpler machine learning potentials and cannot easily handle structures much beyond tens of thousands of atoms. For scientists who need to study rare events, slow diffusion, or complex phase changes, this performance wall turns powerful models into niche tools. The authors aim to keep the benefits of foundation models while stripping away unnecessary cost.
Trimming the neural network without losing its skill
The first step in the proposed workflow is called message-passing depth pruning. The team analyzed how much each layer in several popular atomistic foundation models actually changes the internal description of atoms. They observed an “over-smoothing” effect: after the early layers have done most of the work, later layers only make tiny, repetitive adjustments. Exploiting this, they simply keep the first few layers and remove the rest, then fine-tune the shortened model on task-specific data. Tests on lithium phosphate glass, mixed silicon–silica systems, and metal particles on oxide supports show that even models with only one or two remaining layers can reach nearly the same accuracy as the full networks, while being much lighter and faster. 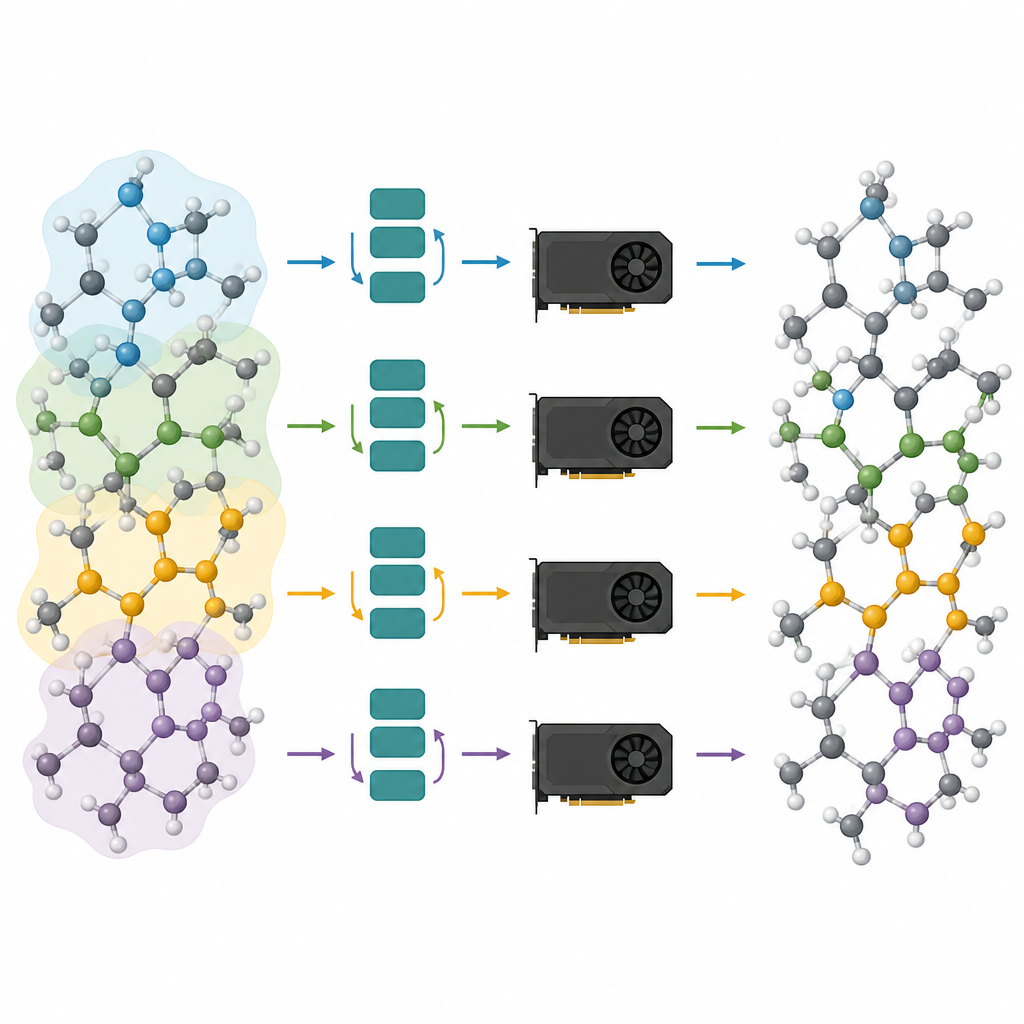
Splitting big atomic systems across many chips
Pruned models are only half of the solution; the other half is running them efficiently on modern graphics processors. The authors view an atomic system as a graph, where atoms are nodes and bonds or near neighbors are links. They partition this graph into overlapping pieces, each with all the neighbors needed for a chosen number of message-passing steps, and assign these subgraphs to different graphics processors. Because the model is now shallow, the required neighborhood is small, and the extra atoms in each piece stay manageable. Crucially, each processor can run its piece independently, with no need to communicate during the heavy computation, and the results are then stitched together. This “halo” style partitioning lets the same method work whether there is a single graphics card or several working in parallel.
What the new tools make possible
By combining pruning with graph partitioning, the authors achieve large gains in speed and scale. They demonstrate that million-atom simulations and even systems with more than five million atoms are feasible on standard hardware, while maintaining the trustworthy forces and structures that foundation models provide. In practical case studies, the pruned models reproduce the formation of amorphous lithium phosphate, nanoscale separation in silicon–silica glasses, and the gradual encapsulation of a platinum nanoparticle by a defective oxide surface. For non-specialists, the message is clear: this work turns previously slow, memory-hungry atomic AIs into lean engines capable of powering realistic materials simulations, opening the door to routine virtual experiments on complex devices and materials.
Citation: Kong, L., Shim, J., Hu, G. et al. Scalable foundation interatomic potentials via message-passing pruning and graph partitioning. npj Comput Mater 12, 180 (2026). https://doi.org/10.1038/s41524-026-02001-4
Keywords: atomistic foundation models, molecular dynamics, graph neural networks, GPU acceleration, materials simulation