Clear Sky Science · es
Potenciales interatómicos fundamentales escalables mediante poda de paso de mensajes y partición de grafos
Por qué importan las películas atómicas más rápidas
Muchas de las tecnologías que nos interesan, desde baterías mejores hasta catalizadores más limpios, dependen de lo que hacen los átomos a lo largo de tiempos largos y a través de enormes números de partículas. Las simulaciones por ordenador pueden crear “películas” de estos átomos en movimiento, pero los métodos más precisos son tan lentos que a menudo se quedan en sistemas diminutos y tiempos muy cortos. Este artículo presenta una forma de conservar la alta precisión de los modelos de inteligencia artificial modernos para átomos al tiempo que reduce su coste tanto que las simulaciones de millones de átomos y nanosegundos de duración se vuelven prácticas en las tarjetas gráficas de hoy. 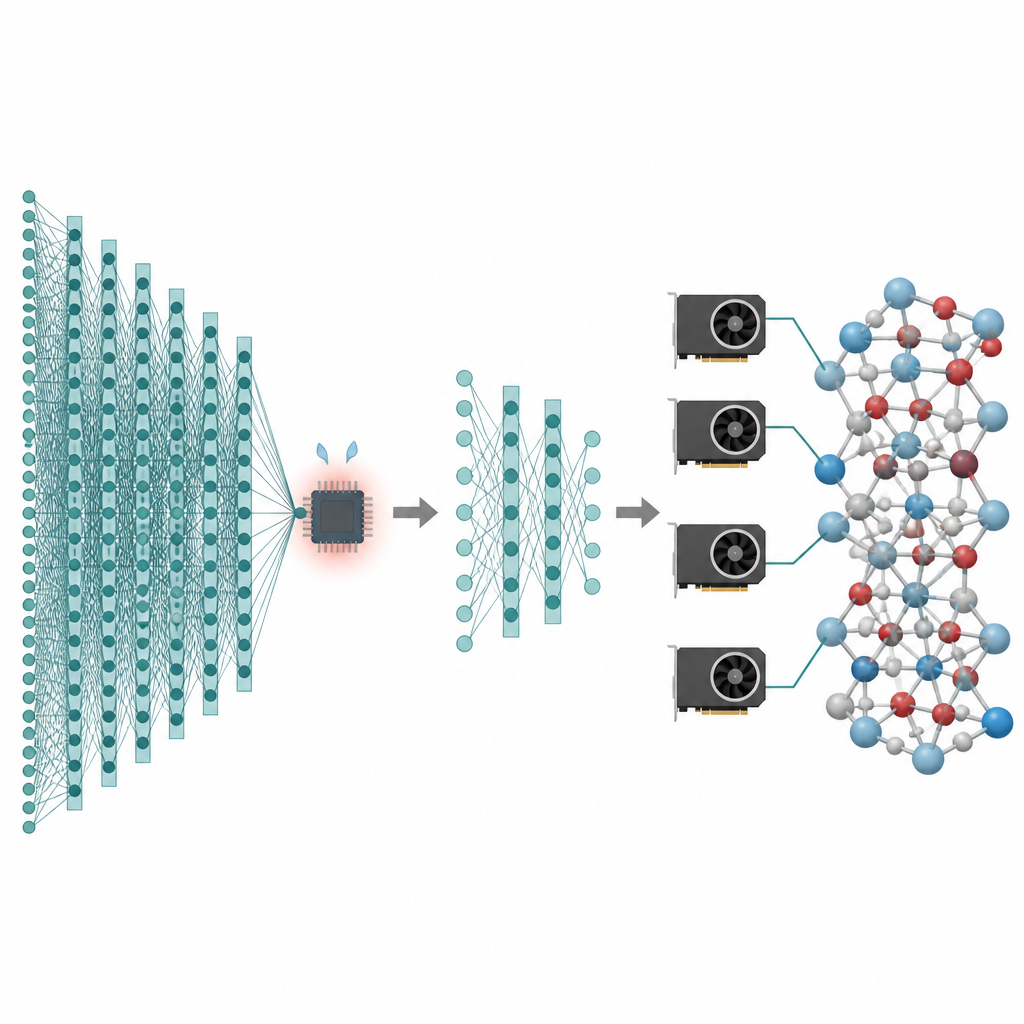
De la precisión cuántica a los límites prácticos
En un extremo, los métodos basados en la mecánica cuántica describen las interacciones atómicas con gran detalle pero son demasiado lentos para sistemas grandes y realistas. Para salvar esta brecha, los investigadores han recurrido a modelos de aprendizaje automático que aprenden cómo se empujan y tiran los átomos entre sí a partir de datos cuánticos, y reemplazan cálculos costosos por predicciones rápidas. Un avance reciente en este campo es la aparición de “modelos fundamentales atomísticos”, grandes redes neuronales entrenadas con colecciones vastas y variadas de estructuras atómicas. Estos modelos pueden ajustarse finamente con solo una pequeña cantidad de datos nuevos y aun así igualar o superar en precisión a muchos modelos hechos a medida, lo que los hace atractivos como herramientas generales para la ciencia de materiales.
Cuando los modelos más grandes se convierten en una carga
La fortaleza de estos modelos fundamentales es también su debilidad. Para cubrir muchos elementos y entornos de enlace, dependen de redes profundas de “paso de mensajes” que intercambian repetidamente información entre átomos vecinos. Cada capa añadida ayuda a los átomos a percibir una región más amplia a su alrededor, pero también infla el tamaño y el uso de memoria del modelo. Como resultado, estos modelos funcionan mucho más despacio que potenciales de aprendizaje automático más simples y no pueden manejar fácilmente estructuras mucho mayores que decenas de miles de átomos. Para los científicos que necesitan estudiar eventos raros, difusión lenta o cambios de fase complejos, este muro de rendimiento convierte a modelos potentes en herramientas de nicho. Los autores pretenden conservar los beneficios de los modelos fundamentales mientras eliminan el coste innecesario.
Recortar la red neuronal sin perder su habilidad
El primer paso en el flujo de trabajo propuesto se denomina poda de profundidad de paso de mensajes. El equipo analizó cuánto cambia cada capa en varios modelos fundamentales atomísticos populares la descripción interna de los átomos. Observaron un efecto de “suavizado excesivo”: después de que las capas iniciales han hecho la mayor parte del trabajo, las capas posteriores sólo hacen ajustes pequeños y repetitivos. Explotando esto, conservan simplemente las primeras capas y eliminan el resto, para luego afinar el modelo acortado con datos específicos de la tarea. Pruebas en vidrio de fosfato de litio, sistemas mixtos silicio–sílice y partículas metálicas sobre soportes oxidados muestran que incluso modelos con sólo una o dos capas restantes pueden alcanzar casi la misma precisión que las redes completas, a la vez que son mucho más ligeros y rápidos. 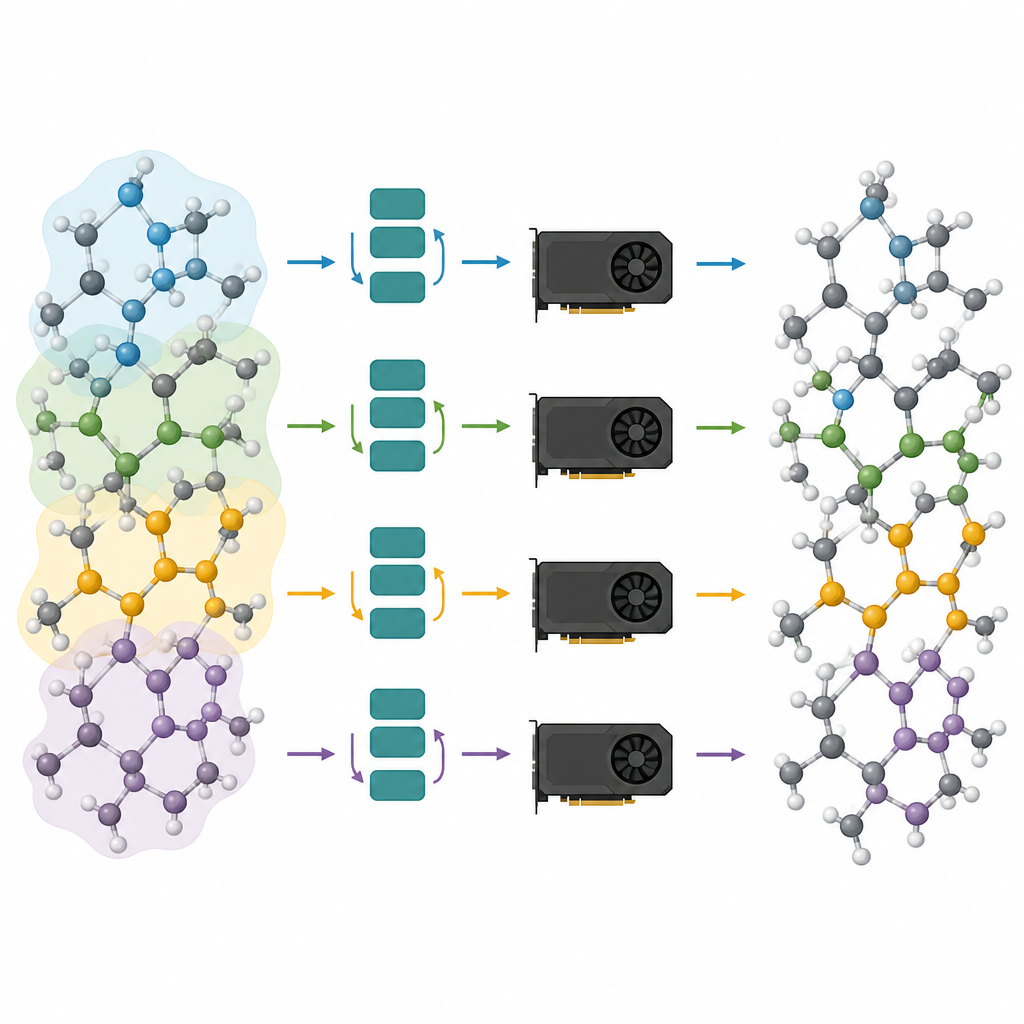
Dividir grandes sistemas atómicos entre muchas tarjetas
Los modelos podados son sólo la mitad de la solución; la otra mitad es ejecutarlos de forma eficiente en las GPUs modernas. Los autores ven un sistema atómico como un grafo, donde los átomos son nodos y los enlaces o vecinos próximos son aristas. Particionan este grafo en piezas superpuestas, cada una con todos los vecinos necesarios para un número elegido de pasos de paso de mensajes, y asignan estos subgrafos a diferentes GPUs. Debido a que el modelo ahora es poco profundo, la vecindad requerida es pequeña y los átomos extra en cada pieza permanecen manejables. De forma crucial, cada procesador puede ejecutar su porción de manera independiente, sin necesidad de comunicarse durante el cálculo pesado, y luego se cosen los resultados. Este tipo de partición con “halo” permite que el mismo método funcione tanto si hay una sola tarjeta gráfica como si varias trabajan en paralelo.
Lo que permiten las nuevas herramientas
Al combinar la poda con la partición de grafos, los autores logran grandes aumentos en velocidad y escala. Demuestran que las simulaciones de millones de átomos e incluso sistemas con más de cinco millones de átomos son factibles en hardware estándar, manteniendo las fuerzas y estructuras confiables que proporcionan los modelos fundamentales. En estudios de caso prácticos, los modelos podados reproducen la formación de fosfato de litio amorfo, la separación a nanoescala en vidrios silicio–sílice y la encapsulación gradual de una nanopartícula de platino por una superficie de óxido defectuosa. Para los no especialistas, el mensaje es claro: este trabajo convierte a las IA atómicas que antes eran lentas y consumidoras de memoria en motores esbeltos capaces de impulsar simulaciones realistas de materiales, abriendo la puerta a experimentos virtuales de rutina sobre dispositivos y materiales complejos.
Cita: Kong, L., Shim, J., Hu, G. et al. Scalable foundation interatomic potentials via message-passing pruning and graph partitioning. npj Comput Mater 12, 180 (2026). https://doi.org/10.1038/s41524-026-02001-4
Palabras clave: modelos fundamentales atomísticos, dinámica molecular, redes neuronales de grafos, aceleración por GPU, simulación de materiales