Clear Sky Science · it
Potenziali interatomici fondamentali scalabili tramite potatura del message-passing e partizionamento del grafo
Perché contano filmati atomici più veloci
Molte delle tecnologie che ci interessano, dalle batterie più performanti ai catalizzatori più puliti, dipendono dal comportamento degli atomi su tempi lunghi e su un numero enorme di particelle. Le simulazioni al computer possono generare “filmati” di questi atomi in movimento, ma i metodi più accurati sono così lenti che spesso si bloccano su sistemi minuscoli e tempi brevissimi. Questo articolo presenta un modo per conservare l’alto livello di accuratezza dei moderni modelli di intelligenza artificiale per atomi riducendone i costi a tal punto che simulazioni di milioni di atomi e della durata di nanosecondi diventano praticabili sulle GPU odierne. 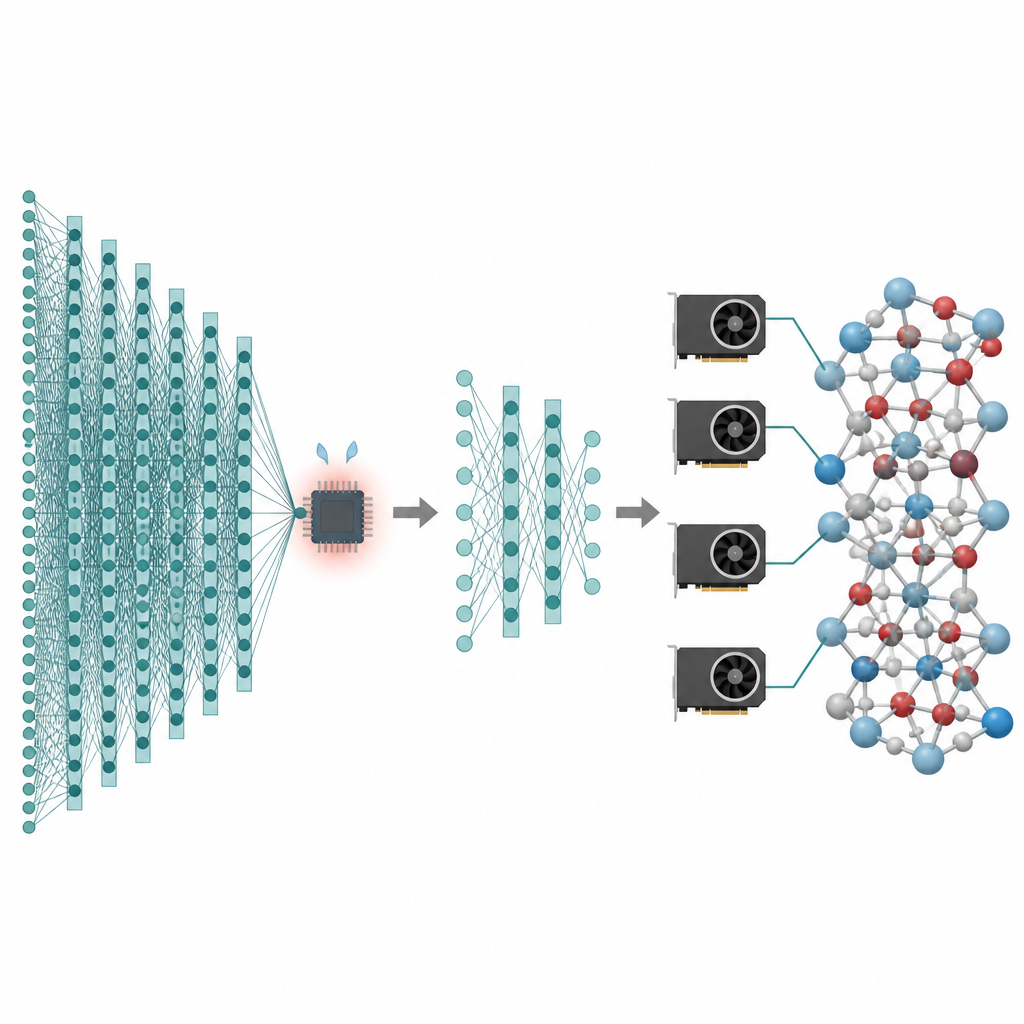
Dall’accuratezza quantistica ai limiti pratici
All’estremo, i metodi basati sulla meccanica quantistica descrivono le interazioni atomiche con grande dettaglio ma sono troppo lenti per sistemi ampi e realistici. Per colmare questo divario, i ricercatori si sono rivolti a modelli di machine learning che apprendono come gli atomi si spingono e si attraggono a partire da dati quantistici, sostituendo i calcoli costosi con previsioni rapide. Un salto recente in questo campo è l’ascesa dei “modelli fondamentali atomistici”, grandi reti neurali addestrate su raccolte vaste e diversificate di strutture atomiche. Questi modelli possono essere rifiniti con una piccola quantità di nuovi dati e riescono comunque a eguagliare o superare molti modelli costruiti su misura in termini di accuratezza, rendendoli strumenti attraenti e generali per la scienza dei materiali.
Quando i modelli più grandi diventano un peso
Il punto di forza di questi modelli fondamentali è anche la loro debolezza. Per coprire molti elementi e ambienti di legame, si basano su profonde reti di «message-passing» che scambiano ripetutamente informazioni tra atomi vicini. Ogni strato aggiunto aiuta gli atomi a percepire una regione più ampia intorno a sé, ma gonfia anche le dimensioni del modello e l’uso di memoria. Di conseguenza, questi modelli girano molto più lentamente rispetto a potenziali di machine learning più semplici e non riescono facilmente a gestire strutture ben oltre le decine di migliaia di atomi. Per gli scienziati che devono studiare eventi rari, diffusione lenta o complesse transizioni di fase, questo limite di prestazioni trasforma modelli potenti in strumenti di nicchia. Gli autori mirano a mantenere i vantaggi dei modelli fondamentali riducendo i costi non necessari.
Potare la rete neurale senza perdere capacità
Il primo passo nel flusso di lavoro proposto si chiama potatura della profondità del message-passing. Il team ha analizzato quanto ogni strato in diversi modelli fondamentali atomistici popolari modifica effettivamente la descrizione interna degli atomi. Hanno osservato un effetto di «over-smoothing»: dopo che i primi strati hanno svolto la maggior parte del lavoro, gli strati successivi effettuano solo piccoli aggiustamenti ripetitivi. Sfruttando questo, hanno mantenuto semplicemente i primi pochi strati e rimosso il resto, quindi hanno rifinito il modello accorciato sui dati specifici del compito. Test su vetro di fosfato di litio, sistemi misti silicio–silice e particelle metalliche su supporti ossidi mostrano che anche modelli con solo uno o due strati residui possono raggiungere quasi la stessa accuratezza delle reti complete, risultando al contempo molto più leggeri e rapidi. 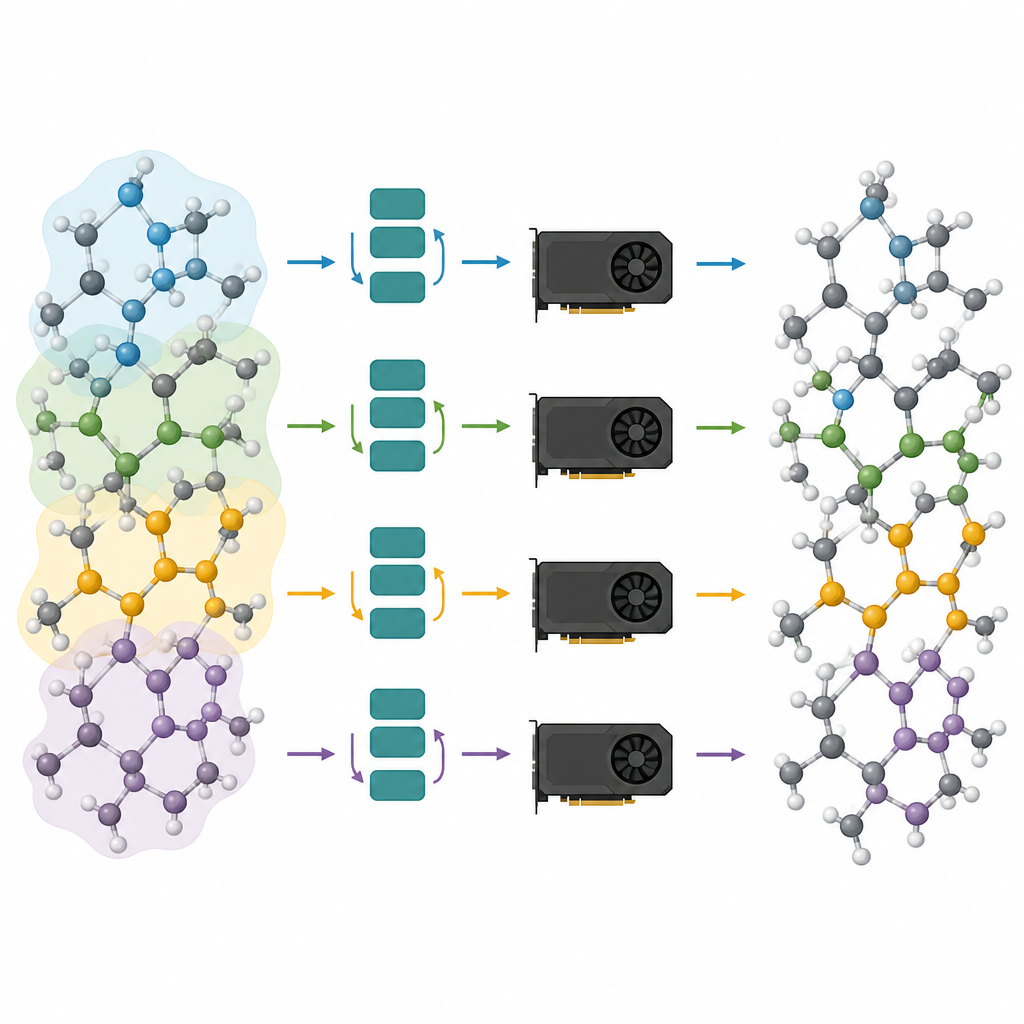
Suddividere grandi sistemi atomici su più chip
I modelli potato sono solo metà della soluzione; l’altra metà consiste nell’eseguirli in modo efficiente sulle moderne GPU. Gli autori considerano un sistema atomico come un grafo, dove gli atomi sono nodi e legami o vicini prossimi sono archi. Partizionano questo grafo in pezzi sovrapposti, ognuno dotato di tutti i vicini necessari per un numero scelto di passi di message-passing, e assegnano questi sottografi a diverse GPU. Poiché il modello è ora superficiale, il vicinato richiesto è piccolo e gli atomi extra in ciascun pezzo rimangono gestibili. In modo cruciale, ogni processore può eseguire il proprio pezzo in modo indipendente, senza necessità di comunicazione durante il calcolo intenso, e i risultati vengono poi ricuciti insieme. Questo partizionamento in stile “alone” permette allo stesso metodo di funzionare sia con una singola scheda grafica sia con più schede in parallelo.
Cosa rendono possibili i nuovi strumenti
Combinando potatura e partizionamento del grafo, gli autori ottengono grandi guadagni in velocità e scala. Dimostrano che simulazioni su milioni di atomi e persino sistemi con oltre cinque milioni di atomi sono fattibili su hardware standard, pur mantenendo le forze e le strutture affidabili che i modelli fondamentali forniscono. In studi di caso pratici, i modelli potato riproducono la formazione di fosfato di litio amorfo, la separazione su scala nanometrica nei vetri silicio–silice e l’incapsulamento graduale di una nanoparticella di platino da parte di una superficie ossida difettosa. Per i non specialisti, il messaggio è chiaro: questo lavoro trasforma AI atomistiche precedentemente lente e voraci di memoria in motori snelli in grado di alimentare simulazioni realistiche dei materiali, aprendo la porta a esperimenti virtuali di routine su dispositivi e materiali complessi.
Citazione: Kong, L., Shim, J., Hu, G. et al. Scalable foundation interatomic potentials via message-passing pruning and graph partitioning. npj Comput Mater 12, 180 (2026). https://doi.org/10.1038/s41524-026-02001-4
Parole chiave: modelli fondamentali atomistici, dinamica molecolare, reti neurali su grafo, accelerazione GPU, simulazione dei materiali