Clear Sky Science · pt
Potenciais interatômicos fundamentais escaláveis via poda de message-passing e particionamento de grafos
Por que filmes atômicos mais rápidos importam
Muitas das tecnologias que nos interessam, desde baterias melhores até catalisadores mais limpos, dependem do comportamento dos átomos ao longo de tempos longos e em enormes quantidades de partículas. Simulações computacionais podem criar “filmes” desses átomos em movimento, mas os métodos mais precisos são tão lentos que frequentemente ficam limitados a sistemas minúsculos e tempos curtíssimos. Este artigo apresenta uma forma de manter a alta precisão dos modelos modernos de inteligência artificial para átomos enquanto reduz tanto seu custo que simulações de milhões de átomos por nanossegundos se tornam práticas nas placas gráficas atuais. 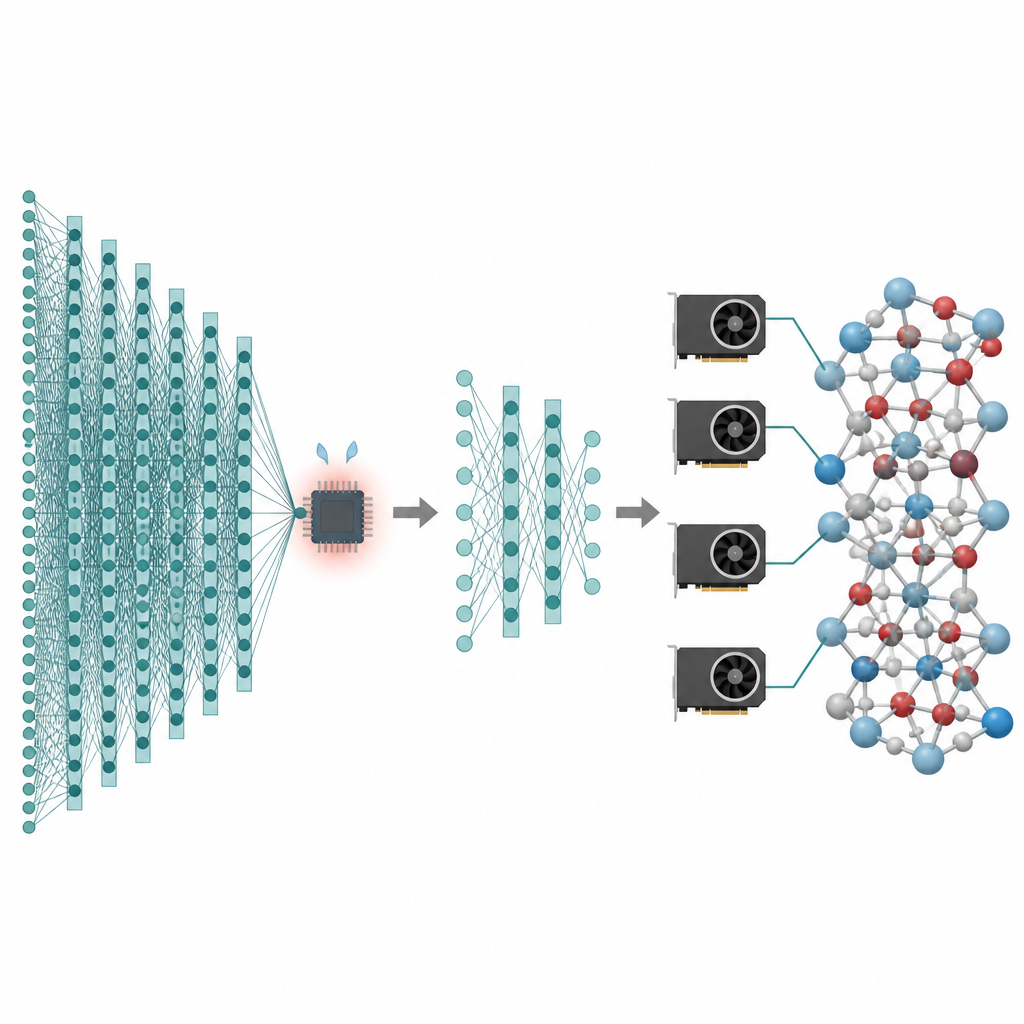
Da precisão quântica aos limites práticos
Em um extremo, métodos baseados em mecânica quântica descrevem as interações atômicas com grande detalhe, mas são muito lentos para sistemas grandes e realistas. Para preencher essa lacuna, pesquisadores recorreram a modelos de aprendizado de máquina que aprendem como os átomos se atraem e se repelem a partir de dados quânticos, substituindo cálculos caros por previsões rápidas. Um avanço recente nessa área é o surgimento dos “modelos fundamentais atomísticos”, grandes redes neurais treinadas em coleções vastas e variadas de estruturas atômicas. Esses modelos podem ser refinados com apenas uma pequena quantidade de novos dados e ainda assim igualar ou superar muitos modelos feitos sob medida em precisão, tornando-os atraentes como ferramentas gerais para a ciência dos materiais.
Quando modelos maiores viram um fardo
A força desses modelos fundamentais é também sua fraqueza. Para cobrir muitos elementos e ambientes de ligação, eles dependem de redes profundas de “message-passing” que trocam repetidamente informação entre átomos vizinhos. Cada camada adicional ajuda os átomos a perceberem uma região mais ampla ao redor, mas também aumenta o tamanho do modelo e o uso de memória. Como resultado, esses modelos rodam muito mais devagar do que potenciais de aprendizado de máquina mais simples e não conseguem lidar facilmente com estruturas além de dezenas de milhares de átomos. Para cientistas que precisam estudar eventos raros, difusão lenta ou mudanças de fase complexas, essa barreira de desempenho transforma modelos poderosos em ferramentas de nicho. Os autores buscam manter os benefícios dos modelos fundamentais ao mesmo tempo em que eliminam custos desnecessários.
Podando a rede neural sem perder sua capacidade
O primeiro passo do fluxo de trabalho proposto chama-se poda da profundidade de message-passing. A equipe analisou quanto cada camada em vários modelos fundamentais atomísticos populares realmente altera a descrição interna dos átomos. Eles observaram um efeito de “over-smoothing”: depois que as camadas iniciais fazem a maior parte do trabalho, camadas posteriores apenas fazem ajustes pequenos e repetitivos. Explorando isso, eles mantêm simplesmente as primeiras camadas e removem o restante, então afinam o modelo encurtado com dados específicos da tarefa. Testes em vidro de fosfato de lítio, sistemas mistos de silício–sílica e partículas metálicas sobre suportes de óxido mostram que mesmo modelos com apenas uma ou duas camadas restantes podem atingir quase a mesma precisão das redes completas, sendo muito mais leves e rápidos. 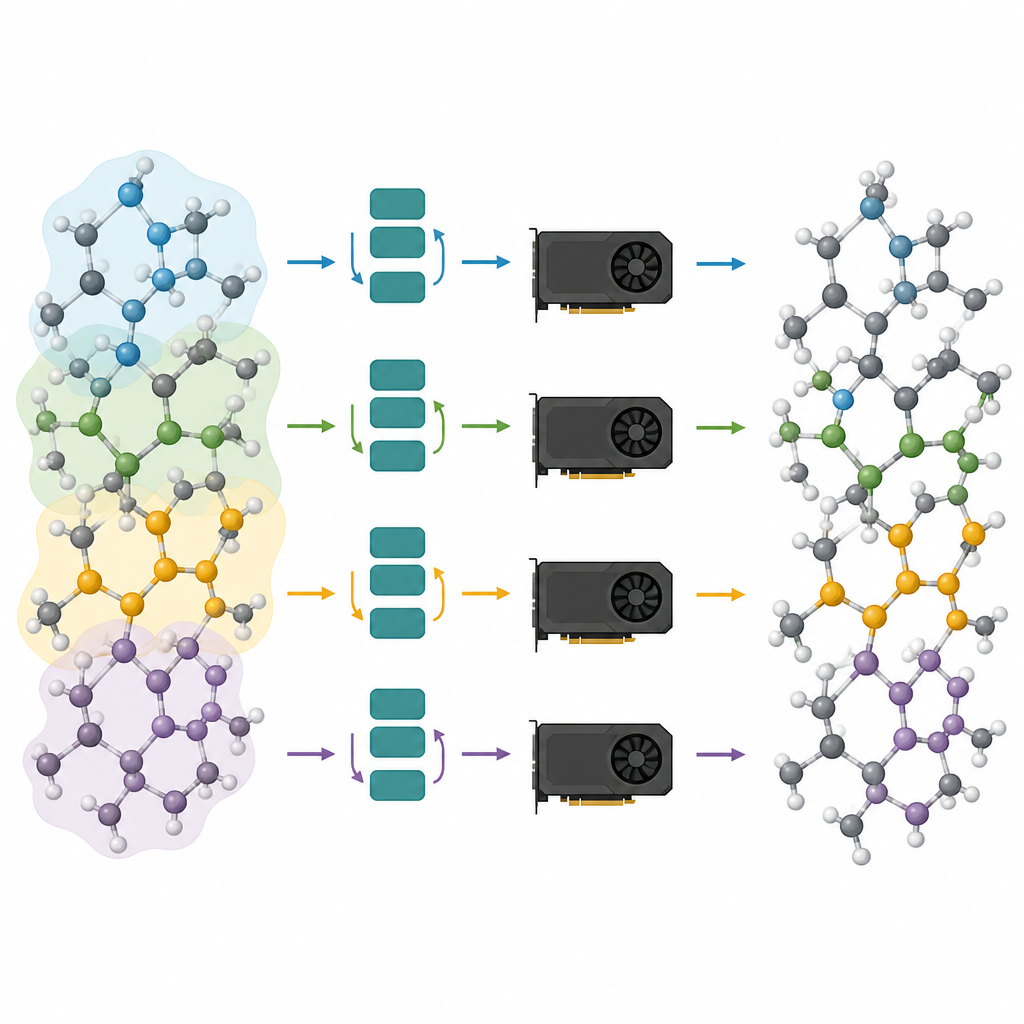
Dividindo grandes sistemas atômicos entre muitos chips
Modelos podados são apenas metade da solução; a outra metade é executá‑los eficientemente em processadores gráficos modernos. Os autores enxergam um sistema atômico como um grafo, onde os átomos são nós e ligações ou vizinhos próximos são arestas. Eles particionam esse grafo em pedaços sobrepostos, cada um contendo todos os vizinhos necessários para um número escolhido de passos de message-passing, e atribuem esses subgrafos a diferentes GPUs. Como o modelo agora é raso, a vizinhança requerida é pequena e os átomos extras em cada pedaço permanecem manejáveis. Crucialmente, cada processador pode rodar seu pedaço de forma independente, sem necessidade de comunicar durante a parte mais custosa do cálculo, e os resultados são então costurados. Esse particionamento em estilo “halo” permite que o mesmo método funcione tanto em uma placa gráfica única quanto em várias trabalhando em paralelo.
O que as novas ferramentas possibilitam
Ao combinar poda com particionamento de grafos, os autores alcançam grandes ganhos em velocidade e escala. Eles demonstram que simulações com milhões de átomos e mesmo sistemas com mais de cinco milhões de átomos são viáveis em hardware padrão, mantendo as forças e estruturas confiáveis que os modelos fundamentais oferecem. Em estudos de caso práticos, os modelos podados reproduzem a formação do fosfato de lítio amorfo, a separação em nanoescala em vidros silício–sílica e a encapsulação gradual de uma nanopartícula de platina por uma superfície de óxido defeituosa. Para não especialistas, a mensagem é clara: este trabalho transforma IAs atômicas anteriormente lentas e famintas por memória em motores enxutos capazes de alimentar simulações realistas de materiais, abrindo a porta para experimentos virtuais de rotina em dispositivos e materiais complexos.
Citação: Kong, L., Shim, J., Hu, G. et al. Scalable foundation interatomic potentials via message-passing pruning and graph partitioning. npj Comput Mater 12, 180 (2026). https://doi.org/10.1038/s41524-026-02001-4
Palavras-chave: modelos fundamentais atomísticos, dinâmica molecular, redes neurais em grafos, aceleração por GPU, simulação de materiais