Clear Sky Science · sv
Upptäckt av dubbelsträngsbrott i DNA av NHEJ-systemet stabiliserar RORγt:s transkriptionella aktivitet och formar Th17-patogenicitet i autoimmunitet
När DNA-reparation förvandlar vänligt beskjutande till sjukdom
Vårt immunförsvar förlitar sig på T‑celler för att stå emot infektioner, men ibland går dessa försvarare rogue och attackerar kroppen själv, vilket orsakar autoimmuna sjukdomar som uveit i ögat eller inflammatorisk tarmsjukdom. Denna studie avslöjar en överraskande vändning: ett DNA‑reparationssystem som normalt skyddar celler från skada kan också fungera som en dold accelerator för en särskilt aggressiv typ av immuncell och bidra till kronisk inflammation istället för att förhindra skada.
DNA‑skada som en dold larmsignal
När T‑celler aktiveras kraftigt, särskilt av själv‑antigener i autoimmuna miljöer, producerar deras mitokondrier vågor av reaktiva syreradikaler som kan orsaka dubbelsträngsbrott i DNA. Normalt är för många sådana brott dödliga. Författarna fann emellertid att en undergrupp kallad patogena Th17‑celler inte bara överlever denna stress utan också utnyttjar den till sin fördel. Dessa celler uppvisar höga nivåer av en DNA‑reparationsväg känd som icke‑homolog ändsammanfogning (NHEJ), som snabbt känner av brott och reagerar. Det handlar inte bara om att lappa DNA: själva upptäckten av brotten verkar föra in signaler i det program som gör Th17‑celler inflammatoriska och sjukdomsframkallande, särskilt i musmodeller för autoimmun uveit och kolit. 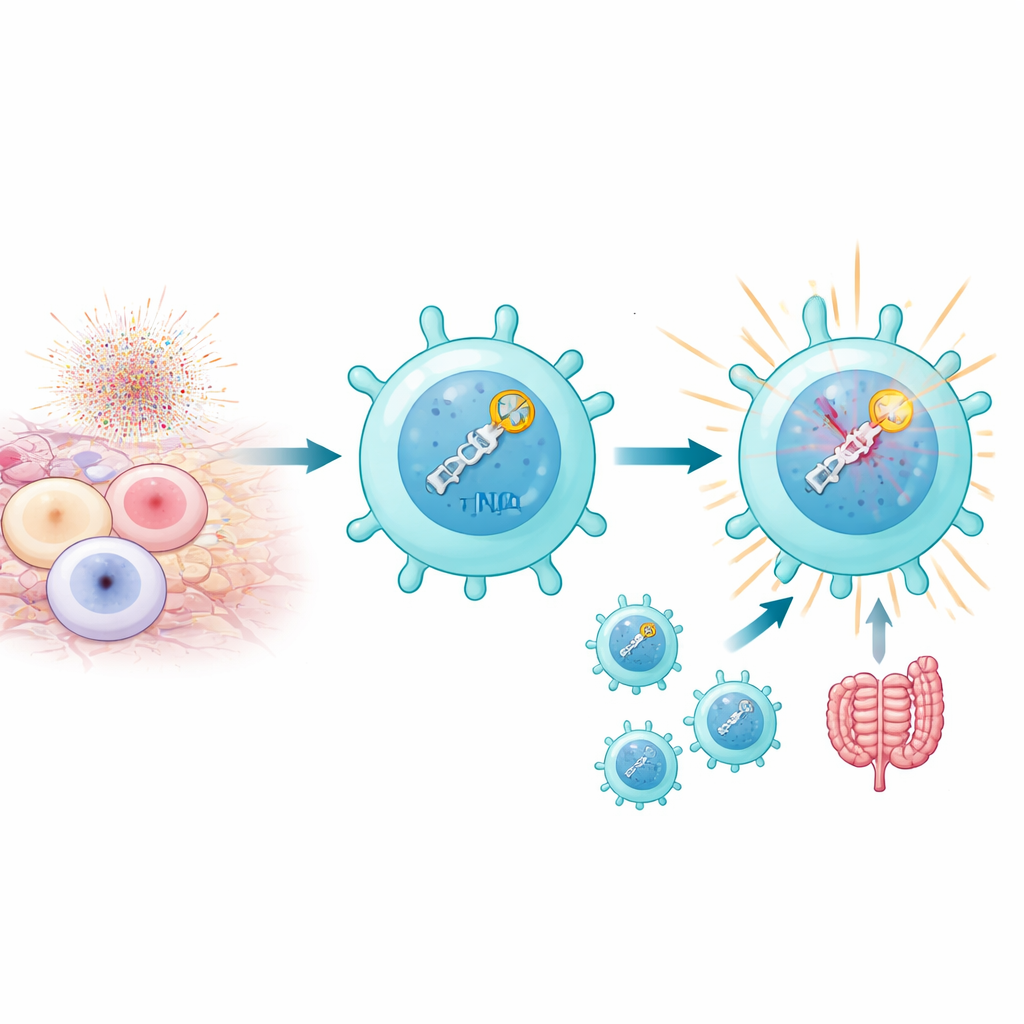
Reparationssensorer, inte reparationen i sig, driver skada
NHEJ är en flerstegsprocess som börjar med sensorproteiner (KU70 och KU80) som fångar in brutna DNA‑ändar och rekryterar ett stort enzym, DNA‑PKcs, som i sin tur tar in ligasproteiner som tätar brottet. Genom att selektivt inaktivera olika delar av denna väg i T‑celler visade forskarna att de tidiga detektionsstegen är avgörande för Th17‑patogenicitet, medan det slutliga ligationssteget överraskande nog är mindre betydelsefullt. Att slå ut KU80 eller DNA‑PKcs minskade kraftigt Th17‑cellernas förmåga att producera inflammatoriska molekyler som IL‑17A, IL‑2 och GM‑CSF och att orsaka vävnadsskada hos möss. Däremot gjorde borttagning av DNA‑ligas IV lite för att dämpa sjukdomen, även om den påverkade cellproliferationen. Dessa experiment indikerar att det som verkligen betyder något för autoimmunitet är själva handlingen att upptäcka DNA‑brott, inte nödvändigtvis att reparera dem.
En DNA‑reparationsenzym agerar som genförstärkare vid sidan om
Vid närmare granskning upptäckte teamet att DNA‑PKcs gör mer än att sitta vid brutna DNA‑ändar. När den aktiveras av KU‑sensorerna modifierar den sig kemiskt i ett område som kallas PQR‑klustret. I detta tillstånd kan DNA‑PKcs fysiskt binda till RORγt, den huvudsakliga transkriptionsfaktorn som definierar Th17‑celler. Detta partnerskap stabiliserar RORγt på DNA nära effektorgen och håller dessa kromatinregioner öppna och tillgängliga. När DNA‑PKcs togs bort, eller när dess PQR‑kluster specifikt raderades, kunde RORγt inte längre inta nyckelplatser vid inflammatoriska gener effektivt och Th17‑celler förlorade mycket av sin kraft att driva autoimmunitet. I musmodeller kunde sådana modifierade celler inte orsaka inflammation i näthinnan trots att de fanns närvarande i immunsystemet. 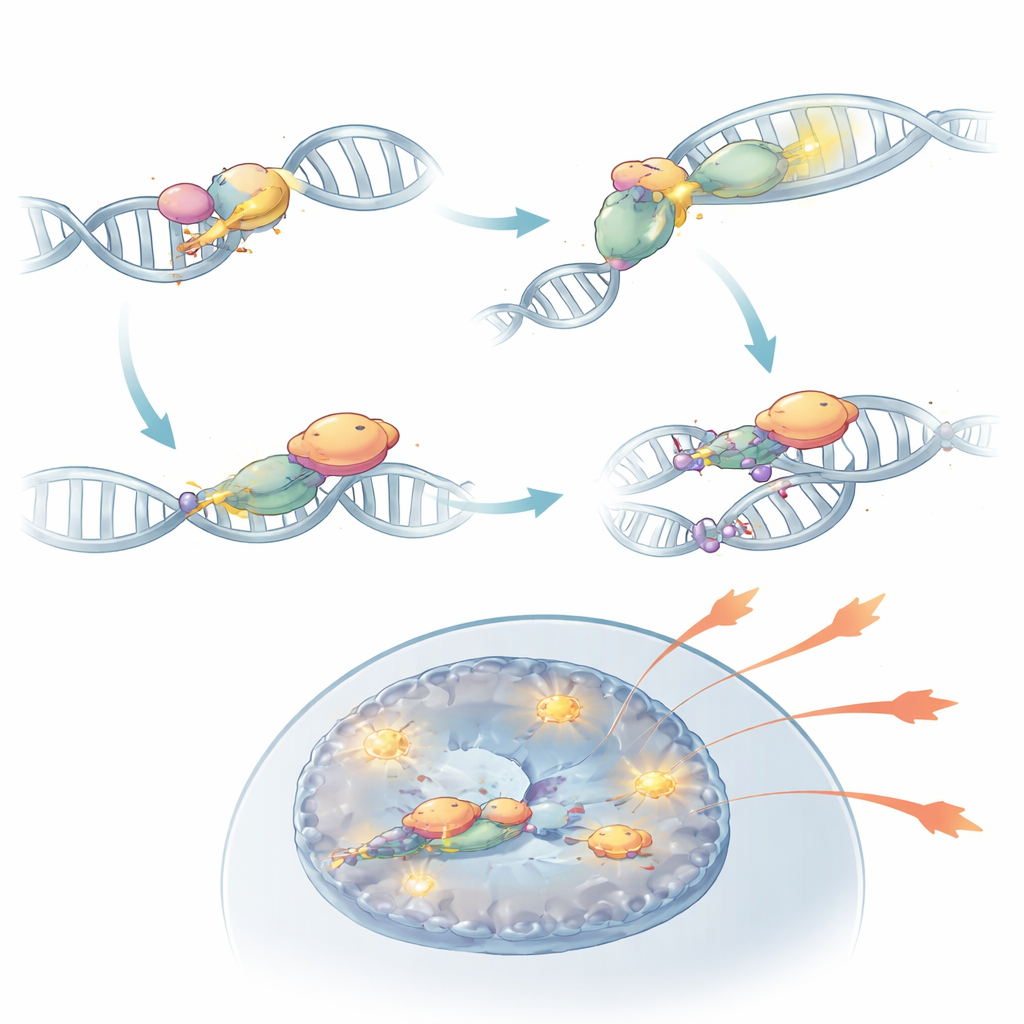
En cytokin‑signal som matar reparation och inflammation
Studien kopplar också detta DNA‑sensor‑nätverk till en välkänd inflammatorisk budbärare, cytokinen IL‑23. IL‑23 är känd för att driva Th17‑celler mot ett skadligt, vävnadsskadande tillstånd. Här visades att IL‑23 ökar NHEJ‑aktivitet, minskar ackumulering av DNA‑brott och förstärker den aktiverande modifieringen av DNA‑PKcs vid PQR‑platsen. Denna effekt var beroende av IL‑23‑receptorn: Th17‑celler som saknade receptorn kunde inte effektivt aktivera NHEJ och samlade på sig mer DNA‑skada. Genom storskaliga protein‑ och RNA‑analyser identifierade författarna ett tidigare underskattat protein, IER2, som en brygga mellan IL‑23‑signalering och DNA‑PKcs. IL‑23 inducerar IER2, som sedan förstärker DNA‑PKcs kinasaktivitet och upprätthåller både DNA‑reparation och RORγt‑driven inflammatorisk genuttryck.
En högrisk‑Th17‑subpopulation hos patienter
För att koppla dessa mekanismer till mänsklig sjukdom profilerade forskarna immunceller från patienter med autoimmun uveit i olika kliniska stadier. Med hjälp av enkelcells‑RNA‑sekvensering upptäckte de en Th17‑subpopulation med höga nivåer av IER2 och NHEJ‑relaterade gener, särskilt komponenter i DNA‑PKcs. Dessa IER2‑höga Th17‑celler var vanligast hos patienter med aktiv, återkommande sjukdom och visade starka signaturer för metabol aktivitet, signalering och inflammation. De bar också markörer för effektiv DNA‑reparation med färre olösta DNA‑brott. Beräkningsbaserade analyser antydde att när Th17‑celler utvecklas längs en bana mot högre IER2‑uttryck, ökar deras inflammatoriska potential parallellt, vilket pekar ut denna subpopulation som en trolig drivkraft för svår autoimmunitet.
Att omvandla ett skyddssystem till ett terapeutiskt mål
Sammanfattningsvis målar arbetet upp en bild där ett DNA‑reparationssensor‑system — avsett att skydda genomet — också fungerar som en kontrollknapp för farliga immunsvar. Patogena Th17‑celler utnyttjar NHEJ‑detektion, DNA‑PKcs‑aktivering och IL‑23–IER2‑axeln för att stabilisera RORγt och bibehålla ett högproducerande inflammatoriskt tillstånd samtidigt som de håller DNA‑skador under kontroll. För patienter antyder detta nya behandlingsvägar: istället för att brett undertrycka immunsystemet skulle läkemedel som selektivt stör KU‑medierad upptäckt av DNA‑brott, DNA‑PKcs‑aktivering vid PQR‑klustret eller IER2–DNA‑PKcs‑interaktionen kunna sänka Th17‑patogenicitet och dämpa autoimmun sjukdom utan att fullständigt blockera väsentlig DNA‑reparation i andra celler.
Citering: Chen, GY., Zhu, WJ., Li, Z. et al. Sensing of DNA double-strand breaks by the NHEJ system stabilizes RORγt transcriptional activity and shapes Th17 pathogenicity in autoimmunity. Cell Res 36, 340–358 (2026). https://doi.org/10.1038/s41422-025-01204-6
Nyckelord: Th17-celler, DNA-reparation, autoimmunitet, DNA-PKcs, IL-23-signalering