Clear Sky Science · pl
Czujność podwójnych pęknięć DNA przez system NHEJ stabilizuje aktywność transkrypcyjną RORγt i kształtuje patogenność Th17 w autoimmunologii
Gdy naprawa DNA zamienia przyjazny ogień w chorobę
Nasz układ odpornościowy polega na limfocytach T, by zwalczać infekcje, ale czasem ci obrońcy wymykają się spod kontroli i atakują własny organizm, wywołując choroby autoimmunologiczne, takie jak zapalenie błony naczyniowej oka czy zapalne choroby jelit. Badanie ujawnia zaskakujący zwrot akcji: system naprawy DNA, który zwykle chroni komórki przed uszkodzeniem, może też działać jako ukryty przyspieszacz szczególnie agresywnego typu komórek odpornościowych, wspomagając przewlekłe zapalenie zamiast zapobiegania mu.
Uszkodzenie DNA jako ukryty sygnał alarmowy
Gdy limfocyty T są silnie aktywowane, szczególnie przez autoantygeny w warunkach autoimmunologicznych, ich mitochondria produkują zryw reaktywnych form tlenu, które mogą łamać DNA na dwuniciowe pęknięcia. Zwykle zbyt wiele takich pęknięć jest śmiertelnych. Autorzy odkryli jednak, że podgrupa nazywana patogennymi komórkami Th17 nie tylko przetrwa ten stres, ale wykorzystuje go na swoją korzyść. Komórki te wykazują wysoką aktywność ścieżki naprawczej DNA znanej jako niehomologiczne łączenie końców (NHEJ), która szybko wykrywa pęknięcia i reaguje. Nie chodzi tu wyłącznie o załatwianie szczelin w DNA: samo wykrywanie pęknięć wydaje się włączać program, który nadaje komórkom Th17 cechy zapalne i chorobotwórcze, szczególnie w modelach mysich zapalenia błony naczyniowej oka i zapalenia jelita. 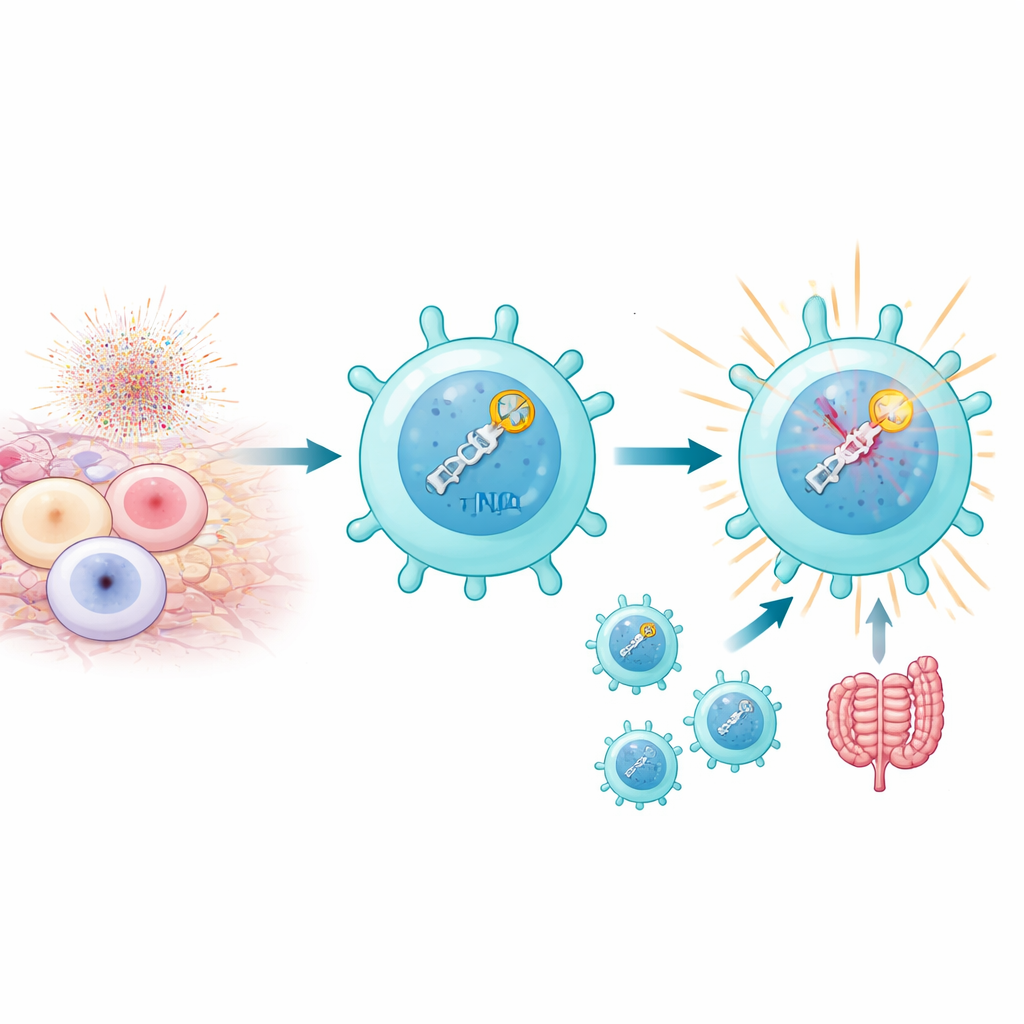
Czujniki naprawy, nie sama naprawa, napędzają szkodę
NHEJ to wieloetapowy proces, zaczynający się od białek-sensorów (KU70 i KU80), które chwytają połamane końce DNA i rekrutują duży enzym DNA-PKcs, który z kolei przyciąga ligazy zamykające przerwę. Poprzez selektywne wyłączanie różnych części tej ścieżki w limfocytach T, badacze pokazali, że wczesne etapy wykrywania są kluczowe dla patogenności Th17, podczas gdy końcowy etap ligacji jest zaskakująco zbędny. Usunięcie KU80 lub DNA-PKcs znacząco zmniejszało zdolność komórek Th17 do wytwarzania cząsteczek zapalnych, takich jak IL-17A, IL-2 i GM-CSF, oraz do powodowania uszkodzeń tkanki u myszy. W przeciwieństwie do tego, usunięcie ligazy DNA IV niewiele zmniejszało nasilenie choroby, choć upośledzało proliferację komórek. Te eksperymenty wskazują, że w autoimmunologii decydujące jest wykrycie pęknięć DNA, a niekoniecznie ich naprawa.
Enzym naprawy DNA pełni drugą rolę jako wzmacniacz genowy
Pogłębiając badania, zespół odkrył, że DNA-PKcs robi więcej niż tylko siedzi na połamanych końcach DNA. Po aktywacji przez sensory KU enzym ten chemicznie modyfikuje sam siebie w regionie zwanym klastrem PQR. W tym stanie DNA-PKcs może fizycznie wiązać się z RORγt, głównym czynnikiem transkrypcyjnym definiującym komórki Th17. To partnerstwo stabilizuje RORγt na DNA w pobliżu genów efektorowych i utrzymuje te regiony chromatyny otwarte i dostępne. Gdy DNA-PKcs został usunięty, albo gdy specyficznie usunięto jego klaster PQR, RORγt nie mógł już skutecznie zajmować kluczowych miejsc genów zapalnych, a komórki Th17 tracily dużą część swojej zdolności do napędzania autoimmunizacji. W modelach mysich takie zmodyfikowane komórki nie powodowały zapalenia siatkówki, mimo że były obecne w układzie odpornościowym. 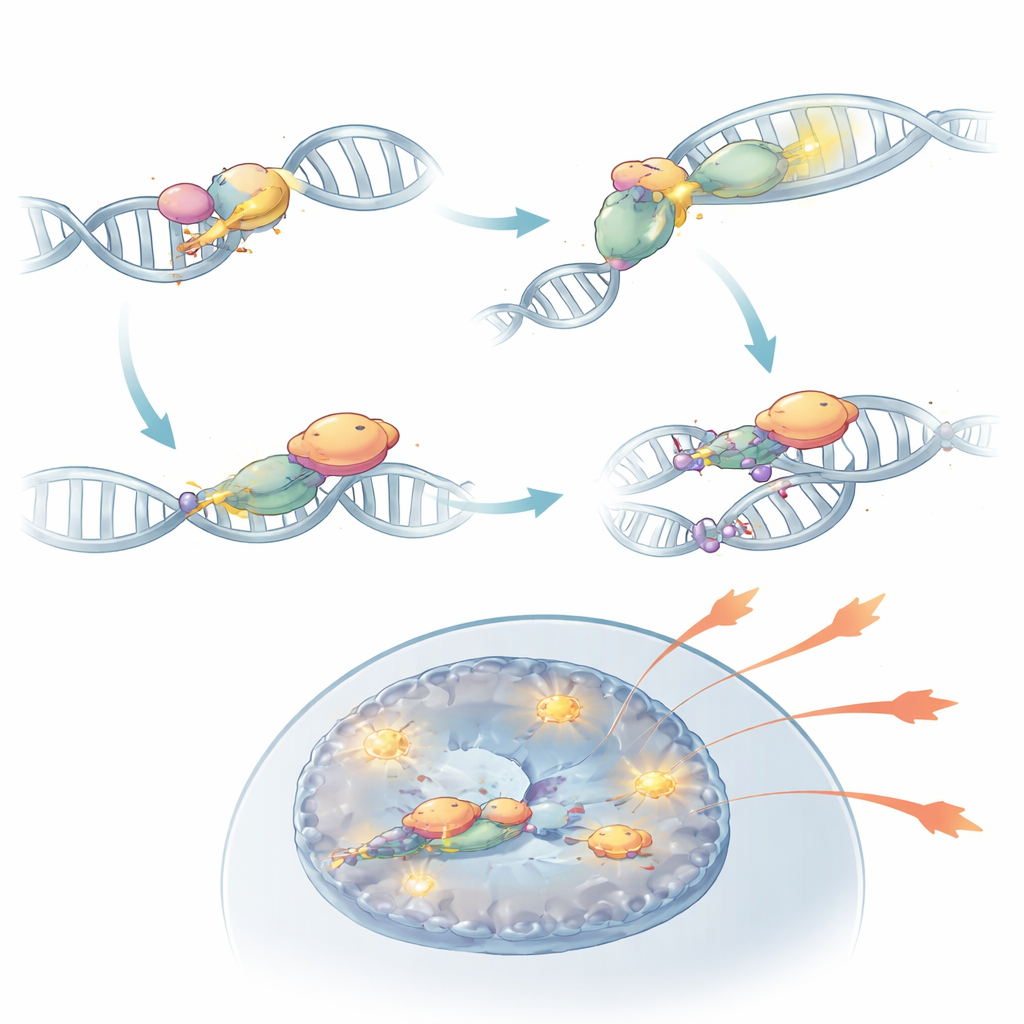
Sygnał cytokinowy, który zasila naprawę i zapalenie
Badanie łączy także tę sieć wykrywania DNA z dobrze znanym mediatorem zapalnym, cytokiną IL-23. IL-23 jest znana z popychania komórek Th17 w kierunku szkodliwego, tkankowo uszkadzającego stanu. Tutaj wykazano, że IL-23 zwiększa aktywność NHEJ, zmniejsza akumulację pęknięć DNA i wzmacnia aktywującą modyfikację DNA-PKcs w miejscu PQR. Efekt ten zależał od receptora IL-23: komórki Th17 pozbawione receptora nie były w stanie efektywnie aktywować NHEJ i gromadziły więcej uszkodzeń DNA. Dzięki analizom białkowymi i RNA na dużą skalę autorzy zidentyfikowali wcześniej niedoceniane białko IER2 jako most łączący sygnalizację IL-23 z DNA-PKcs. IL-23 indukuje IER2, które następnie wzmacnia kinazową aktywność DNA-PKcs, podtrzymując zarówno naprawę DNA, jak i ekspresję zapalnych genów zależnych od RORγt.
Wysokiego ryzyka podzbiór Th17 u pacjentów
Aby powiązać te mechanizmy z ludzką chorobą, badacze profilowali komórki odpornościowe od pacjentów z autoimmunologicznym zapaleniem błony naczyniowej oka na różnych etapach choroby. Przy użyciu sekwencjonowania RNA pojedynczych komórek odkryli podpopulację Th17 o wysokim poziomie IER2 i genów związanych z NHEJ, w szczególności składników DNA-PKcs. Komórki Th17 z wysokim IER2 były najliczniejsze u pacjentów z aktywną, nawrotową chorobą i prezentowały silne sygnatury aktywności metabolicznej, sygnalizacji i zapalenia. Miały też markery efektywnej naprawy DNA i mniej nierozwiązanych pęknięć DNA. Analizy komputerowe sugerowały, że w miarę jak komórki Th17 rozwijają się w kierunku wyższej ekspresji IER2, ich potencjał zapalny rośnie równolegle, co wskazuje na tę podpopulację jako prawdopodobnego sprawcę ciężkiej autoimmunizacji.
Przekształcenie systemu ochronnego w cel terapeutyczny
Ogólnie praca kreśli obraz, w którym system sensorów naprawy DNA — mający za zadanie chronić genom — pełni także rolę regulatora niebezpiecznych reakcji immunologicznych. Patogenne komórki Th17 wykorzystują wykrywanie NHEJ, aktywację DNA-PKcs i oś IL-23–IER2 do stabilizacji RORγt i utrzymania wysokiego poziomu stanu zapalnego, jednocześnie kontrolując uszkodzenia DNA. Dla pacjentów sugeruje to nowe strategie terapeutyczne: zamiast szeroko tłumić układ odpornościowy, leki selektywnie zakłócające wykrywanie pęknięć DNA przez KU, aktywację DNA-PKcs w klastrze PQR lub interakcję IER2–DNA-PKcs mogłyby zmniejszyć patogenność Th17 i złagodzić choroby autoimmunologiczne bez całkowitego blokowania niezbędnej naprawy DNA w innych komórkach.
Cytowanie: Chen, GY., Zhu, WJ., Li, Z. et al. Sensing of DNA double-strand breaks by the NHEJ system stabilizes RORγt transcriptional activity and shapes Th17 pathogenicity in autoimmunity. Cell Res 36, 340–358 (2026). https://doi.org/10.1038/s41422-025-01204-6
Słowa kluczowe: komórki Th17, naprawa DNA, autoimmunizacja, DNA-PKcs, sygnalizacja IL-23