Clear Sky Science · es
La detección de roturas de doble cadena del ADN por el sistema NHEJ estabiliza la actividad transcripcional de RORγt y conforma la patogenicidad Th17 en la autoinmunidad
Cuando la reparación del ADN convierte el fuego amigo en enfermedad
Nuestro sistema inmune depende de las células T para repeler infecciones, pero a veces esos defensores se vuelven rebeldes y atacan el propio organismo, provocando enfermedades autoinmunes como la uveítis ocular o la enfermedad inflamatoria intestinal. Este estudio revela un giro sorprendente: un sistema de reparación del ADN que normalmente protege a las células frente al daño también puede actuar como un acelerador oculto para un tipo particularmente agresivo de célula inmunitaria, impulsando la inflamación crónica en lugar de prevenir el daño.
El daño al ADN como señal de alarma oculta
Cuando las células T se activan fuertemente, especialmente por autoantígenos en contextos autoinmunes, sus mitocondrias generan estallidos de especies reactivas de oxígeno que pueden provocar roturas de doble cadena en el ADN. Normalmente, demasiadas de estas roturas son letales. Sin embargo, los autores observaron que un subconjunto llamado células Th17 patógenas no solo sobrevive a este estrés, sino que lo aprovecha. Estas células presentan niveles altos de una vía de reparación del ADN conocida como unión por extremos no homólogos (NHEJ), que detecta rápidamente las roturas y responde. Esto no se reduce solo a parchear el ADN: la detección de las roturas en sí parece alimentarse del programa que convierte a las Th17 en células inflamatorias y causantes de enfermedad, particularmente en modelos murinos de uveítis autoinmune y colitis. 
Sensores de reparación, no la reparación en sí, impulsan el daño
NHEJ es un proceso en múltiples pasos, que comienza con proteínas sensoras (KU70 y KU80) que se adhieren a los extremos rotos del ADN y reclutan una enzima grande, DNA-PKcs, que a su vez incorpora ligasas que sellan la rotura. Al desactivar selectivamente distintas partes de esta vía en células T, los investigadores demostraron que los pasos iniciales de detección son cruciales para la patogenicidad Th17, mientras que el paso final de ligación resulta sorprendentemente prescindible. Eliminar KU80 o DNA-PKcs redujo drásticamente la capacidad de las Th17 para producir moléculas inflamatorias como IL-17A, IL-2 y GM-CSF y para causar daño tisular en ratones. En contraste, suprimir la ligasa IV del ADN hizo poco por mitigar la enfermedad, aunque afectó la proliferación celular. Estos experimentos indican que lo que realmente importa para la autoinmunidad es el acto de detectar las roturas de ADN, no necesariamente repararlas.
Una enzima de reparación del ADN actúa también como potenciador génico
Al profundizar, el equipo descubrió que DNA-PKcs hace más que permanecer en los extremos rotos del ADN. Una vez activada por los sensores KU, se modifica químicamente en una región llamada conglomerado PQR. En este estado, DNA-PKcs puede unirse físicamente a RORγt, el factor de transcripción maestro que define las células Th17. Esta asociación estabiliza a RORγt en el ADN cerca de genes efectoras y mantiene esas regiones de la cromatina abiertas y accesibles. Cuando se eliminó DNA-PKcs, o se borró específicamente su conglomerado PQR, RORγt ya no pudo ocupar eficazmente sitios clave de genes inflamatorios, y las Th17 perdieron gran parte de su capacidad para alimentar la autoinmunidad. En modelos murinos, dichas células modificadas no llegaron a inflamar la retina pese a estar presentes en el sistema inmune. 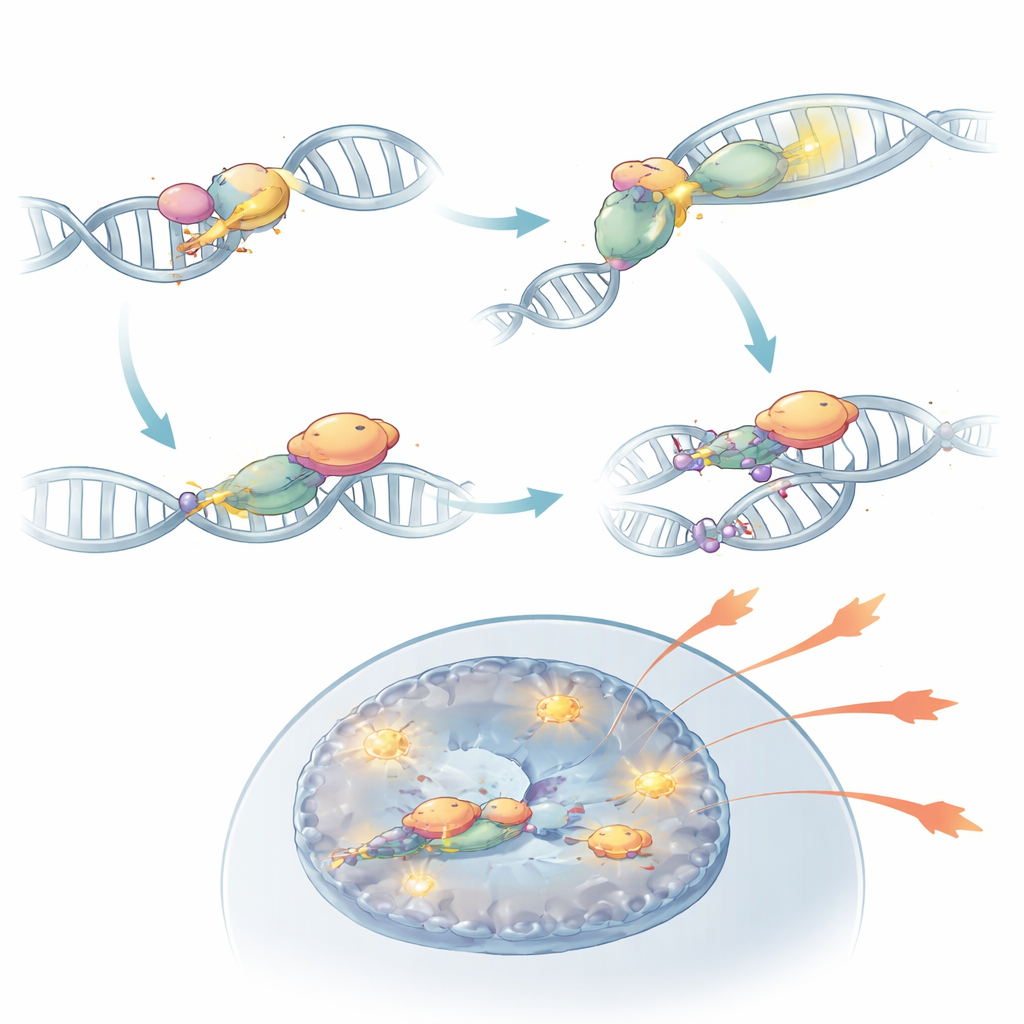
Una señal de citocina que alimenta la reparación y la inflamación
El estudio también vincula esta red de detección del ADN con un mensajero inflamatorio bien conocido, la citocina IL-23. Se sabe que IL-23 empuja a las Th17 hacia un estado dañino para los tejidos. Aquí, se mostró que IL-23 incrementa la actividad de NHEJ, reduce la acumulación de roturas de ADN y potencia la modificación activadora de DNA-PKcs en el sitio PQR. Este efecto dependía del receptor de IL-23: las Th17 sin el receptor no podían activar NHEJ de forma eficiente y acumulaban más daño en el ADN. Mediante análisis proteómicos y de ARN a gran escala, los autores identificaron una proteína hasta ahora poco apreciada, IER2, como un puente entre la señalización de IL-23 y DNA-PKcs. IL-23 induce IER2, que a su vez potencia la actividad quinasa de DNA-PKcs, sosteniendo tanto la reparación del ADN como la expresión génica inflamatoria dirigida por RORγt.
Un subconjunto Th17 de alto riesgo en pacientes
Para vincular estos mecanismos con la enfermedad humana, los investigadores perfilizaron células inmunitarias de pacientes con uveítis autoinmune en distintas etapas clínicas. Usando secuenciación de ARN de célula única, descubrieron una subpoblación de Th17 con altos niveles de IER2 y genes relacionados con NHEJ, en particular componentes de DNA-PKcs. Estas Th17 con IER2 elevado eran más abundantes en pacientes con enfermedad activa y recidivante y mostraban fuertes firmas de actividad metabólica, señalización e inflamación. También presentaban marcadores de reparación del ADN eficiente con menos roturas no resueltas. El análisis computacional sugirió que, a medida que las Th17 avanzan a lo largo de una trayectoria de desarrollo hacia mayor expresión de IER2, su potencial inflamatorio aumenta en paralelo, marcando a este subconjunto como probable impulsor de autoinmunidad severa.
Convertir un sistema protector en un objetivo terapéutico
En conjunto, el trabajo dibuja un panorama en el que un sistema sensor de reparación del ADN—destinado a salvaguardar el genoma—también funciona como un regulador de respuestas inmunes peligrosas. Las Th17 patógenas aprovechan la detección por NHEJ, la activación de DNA-PKcs y el eje IL-23–IER2 para estabilizar RORγt y mantener un estado inflamatorio de alta producción mientras mantienen el daño del ADN bajo control. Para los pacientes, esto sugiere nuevas vías terapéuticas: en lugar de suprimir ampliamente el sistema inmune, fármacos que interfieran selectivamente con la detección de roturas mediada por KU, la activación de DNA-PKcs en el conglomerado PQR o la interacción IER2–DNA-PKcs podrían reducir la patogenicidad Th17 y calmar la enfermedad autoinmune sin bloquear por completo la reparación esencial del ADN en otras células.
Cita: Chen, GY., Zhu, WJ., Li, Z. et al. Sensing of DNA double-strand breaks by the NHEJ system stabilizes RORγt transcriptional activity and shapes Th17 pathogenicity in autoimmunity. Cell Res 36, 340–358 (2026). https://doi.org/10.1038/s41422-025-01204-6
Palabras clave: células Th17, reparación del ADN, autoinmunidad, DNA-PKcs, señalización IL-23