Clear Sky Science · en
Sensing of DNA double-strand breaks by the NHEJ system stabilizes RORγt transcriptional activity and shapes Th17 pathogenicity in autoimmunity
When DNA Repair Turns Friendly Fire into Disease
Our immune system relies on T cells to fend off infections, but sometimes these defenders go rogue and attack the body itself, causing autoimmune diseases such as uveitis in the eye or inflammatory bowel disease. This study reveals a surprising twist: a DNA repair system that normally protects cells from damage can also act as a hidden accelerator for a particularly aggressive type of immune cell, helping drive chronic inflammation instead of preventing harm.
DNA Damage as a Hidden Alarm Signal
When T cells are strongly activated, especially by self-antigens in autoimmune settings, their mitochondria produce bursts of reactive oxygen species that can snap DNA into double-strand breaks. Normally, too many of these breaks are lethal. Yet the authors found that a subset called pathogenic Th17 cells not only survives this stress but uses it to its advantage. These cells carry high levels of a DNA repair pathway known as non-homologous end joining (NHEJ), which rapidly senses breaks and responds. This is not just about patching DNA: the sensing of breaks itself appears to feed into the program that makes Th17 cells inflammatory and disease-causing, particularly in mouse models of autoimmune uveitis and colitis. 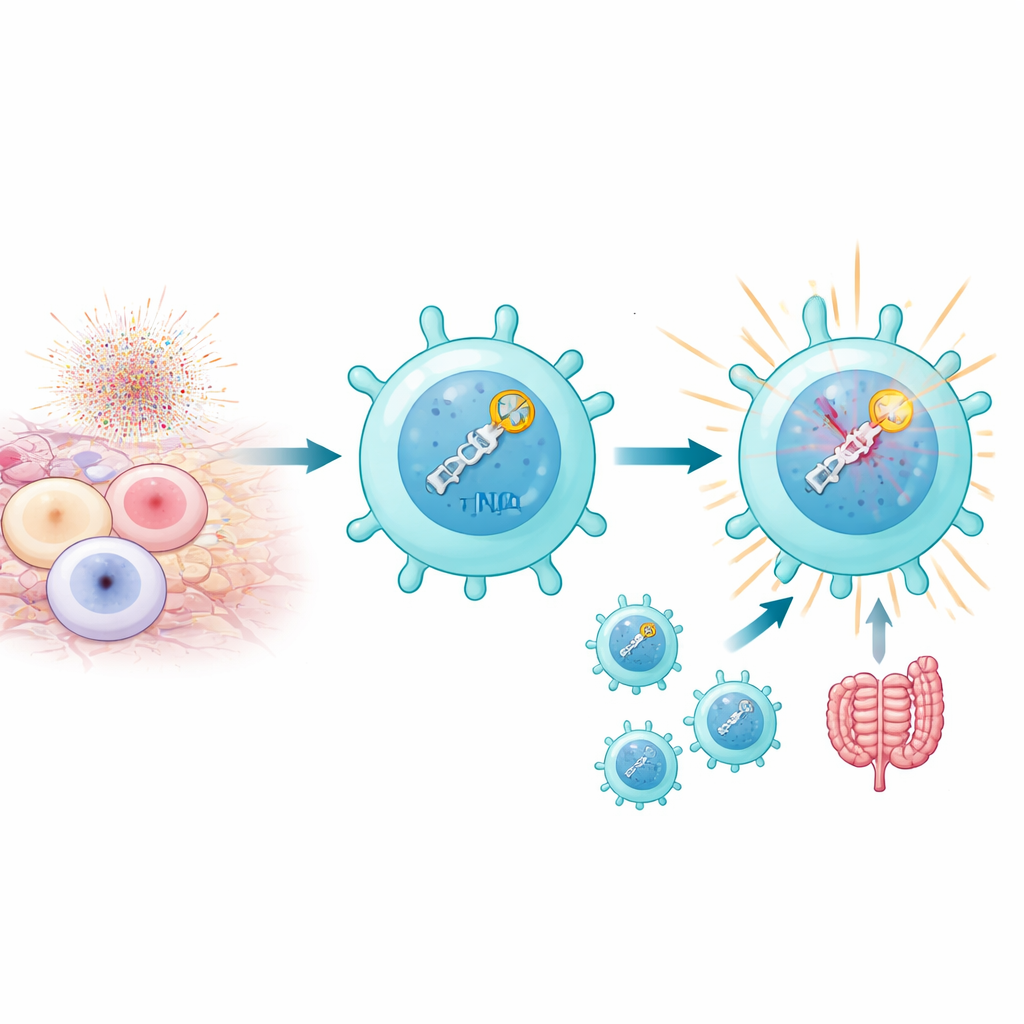
Repair Sensors, Not Repair Itself, Drive Harm
NHEJ is a multi-step process, starting with sensor proteins (KU70 and KU80) that grab broken DNA ends and recruit a large enzyme, DNA-PKcs, which in turn brings in ligase proteins that seal the break. By selectively disabling different parts of this pathway in T cells, the researchers showed that the early sensing steps are crucial for Th17 pathogenicity, while the final ligation step is surprisingly dispensable. Knocking out KU80 or DNA-PKcs sharply reduced the ability of Th17 cells to produce inflammatory molecules such as IL-17A, IL-2, and GM-CSF and to cause tissue damage in mice. In contrast, removing DNA ligase IV did little to blunt disease, even though it impaired cell proliferation. These experiments indicate that what really matters for autoimmunity is the act of detecting DNA breaks, not necessarily fixing them.
A DNA Repair Enzyme Moonlights as a Gene Booster
Diving deeper, the team discovered that DNA-PKcs does more than sit at broken DNA ends. Once activated by the KU sensors, it chemically modifies itself at a region called the PQR cluster. In this state, DNA-PKcs can physically bind to RORγt, the master transcription factor that defines Th17 cells. This partnership stabilizes RORγt on the DNA near effector genes and keeps those regions of chromatin open and accessible. When DNA-PKcs was removed, or when its PQR cluster was specifically deleted, RORγt could no longer occupy key inflammatory gene sites effectively, and Th17 cells lost much of their power to fuel autoimmunity. In mouse models, such modified cells failed to inflame the retina despite being otherwise present in the immune system. 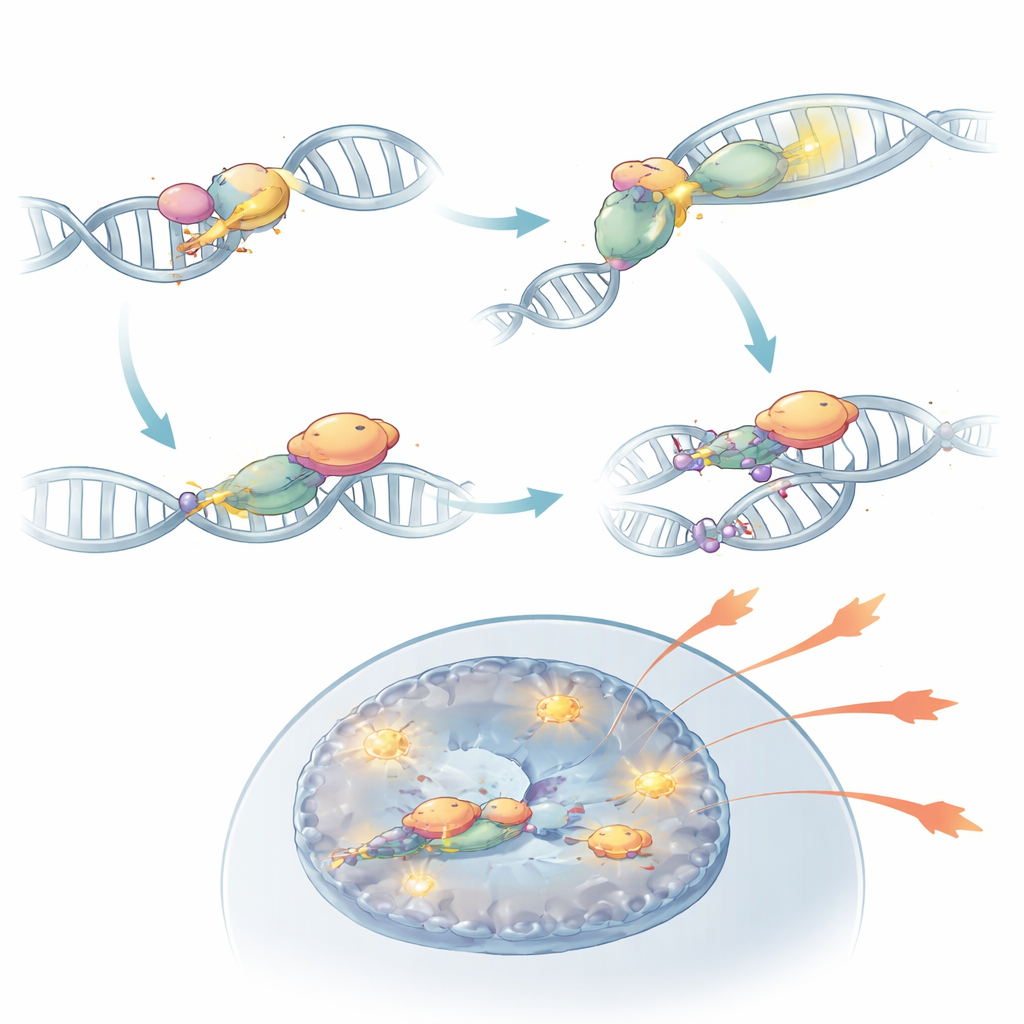
A Cytokine Signal that Feeds Repair and Inflammation
The study also links this DNA sensing network to a well-known inflammatory messenger, the cytokine IL-23. IL-23 is known to push Th17 cells toward a harmful, tissue-damaging state. Here, IL-23 was shown to ramp up NHEJ activity, reduce DNA break accumulation, and enhance the activating modification of DNA-PKcs at the PQR site. This effect depended on the IL-23 receptor: Th17 cells lacking the receptor could not efficiently activate NHEJ and accumulated more DNA damage. Through large-scale protein and RNA analyses, the authors identified a previously underappreciated protein, IER2, as a bridge between IL-23 signaling and DNA-PKcs. IL-23 induces IER2, which then boosts DNA-PKcs’s kinase activity, sustaining both DNA repair and RORγt-driven inflammatory gene expression.
A High-Risk Th17 Subset in Patients
To connect these mechanisms to human disease, the researchers profiled immune cells from patients with autoimmune uveitis at different clinical stages. Using single-cell RNA sequencing, they uncovered a Th17 subpopulation with high levels of IER2 and NHEJ-related genes, particularly DNA-PKcs components. These IER2-high Th17 cells were most abundant in patients with active, relapsing disease and showed strong signatures of metabolic activity, signaling, and inflammation. They also bore markers of efficient DNA repair with fewer unresolved DNA breaks. Computational analysis suggested that as Th17 cells progress along a developmental trajectory toward higher IER2 expression, their inflammatory potential rises in parallel, marking this subset as a likely driver of severe autoimmunity.
Turning a Protective System into a Therapeutic Target
Overall, the work paints a picture in which a DNA repair sensor system—meant to safeguard the genome—doubles as a control knob for dangerous immune responses. Pathogenic Th17 cells leverage NHEJ sensing, DNA-PKcs activation, and the IL-23–IER2 axis to stabilize RORγt and maintain a high-output inflammatory state while keeping DNA damage in check. For patients, this suggests new treatment angles: rather than broadly suppressing the immune system, drugs that selectively interfere with KU-mediated DNA break sensing, DNA-PKcs activation at the PQR cluster, or the IER2–DNA-PKcs interaction could dial down Th17 pathogenicity and calm autoimmune disease without completely blocking essential DNA repair in other cells.
Citation: Chen, GY., Zhu, WJ., Li, Z. et al. Sensing of DNA double-strand breaks by the NHEJ system stabilizes RORγt transcriptional activity and shapes Th17 pathogenicity in autoimmunity. Cell Res 36, 340–358 (2026). https://doi.org/10.1038/s41422-025-01204-6
Keywords: Th17 cells, DNA repair, autoimmunity, DNA-PKcs, IL-23 signaling