Clear Sky Science · fr
La détection des cassures doubles de l’ADN par le système NHEJ stabilise l’activité transcriptionnelle de RORγt et façonne la pathogénicité des Th17 dans l’auto-immunité
Quand la réparation de l’ADN transforme le tir ami en maladie
Notre système immunitaire s’appuie sur les cellules T pour combattre les infections, mais parfois ces défenseurs dérapent et attaquent l’organisme lui‑même, provoquant des maladies auto‑immunes comme l’uvéite oculaire ou la maladie inflammatoire de l’intestin. Cette étude révèle un retournement surprenant : un système de réparation de l’ADN qui protège normalement les cellules contre les dommages peut aussi jouer le rôle d’accélérateur caché pour un type particulièrement agressif de cellules immunitaires, contribuant à entretenir l’inflammation chronique plutôt qu’à prévenir les dégâts.
Les dommages de l’ADN comme signal d’alarme caché
Lorsque les cellules T sont fortement activées, notamment par des auto‑antigènes dans des contextes auto‑immuns, leurs mitochondries produisent des rafales d’espèces réactives de l’oxygène qui peuvent rompre l’ADN en cassures doubles brins. En général, un excès de ces cassures est létal. Pour autant, les auteurs ont constaté qu’un sous‑ensemble appelé Th17 pathogéniques non seulement survit à ce stress mais en tire avantage. Ces cellules présentent des niveaux élevés d’une voie de réparation de l’ADN connue sous le nom de jonction d’extrémités non homologues (NHEJ), qui détecte rapidement les cassures et y répond. Il ne s’agit pas seulement de colmater l’ADN : la détection des cassures semble alimenter le programme qui rend les Th17 inflammatoires et responsables de la maladie, en particulier dans des modèles murins d’uvéite auto‑immune et de colite. 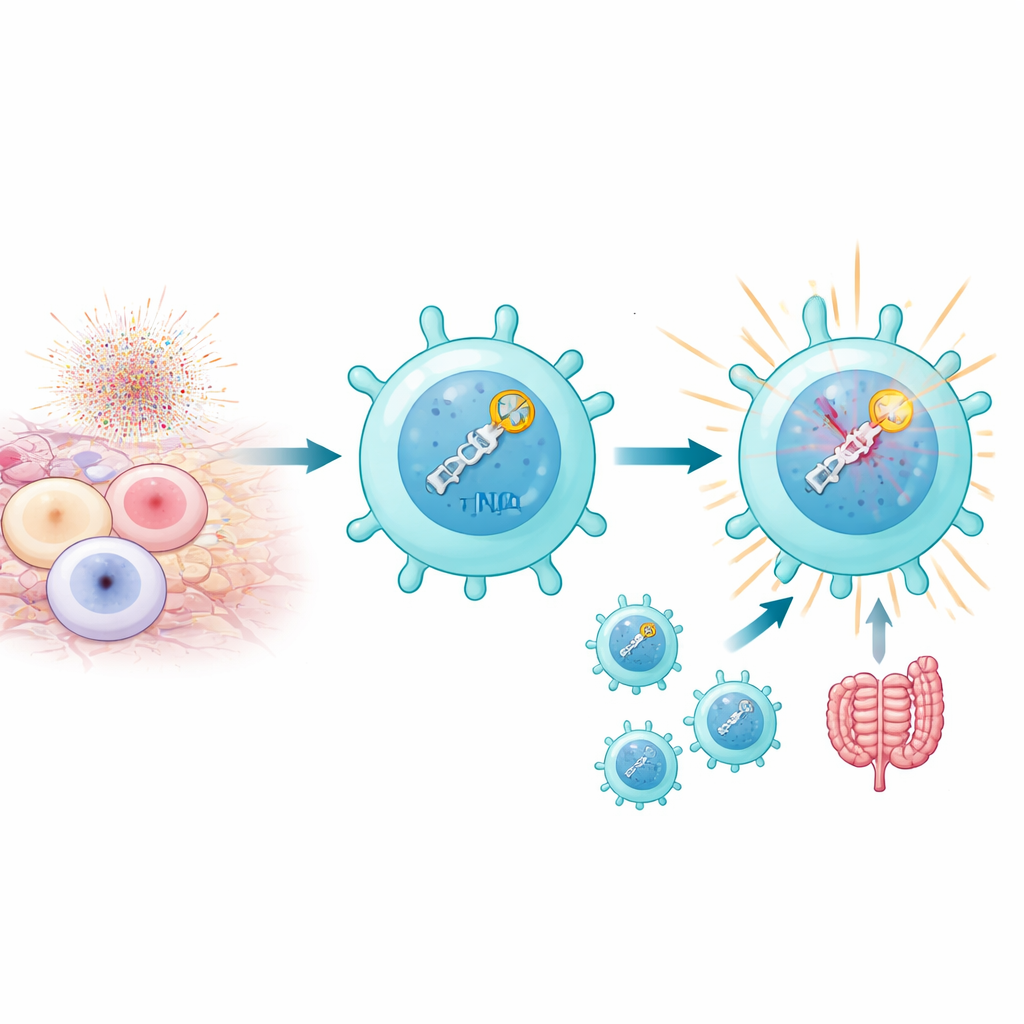
Ce sont les capteurs, pas la réparation finale, qui causent le tort
La NHEJ est un processus en plusieurs étapes, commençant par des protéines détectrices (KU70 et KU80) qui saisissent les extrémités d’ADN brisées et recrutent une grosse enzyme, DNA‑PKcs, laquelle fait ensuite venir des ligases qui scellent la cassure. En désactivant sélectivement différentes parties de cette voie dans les cellules T, les chercheurs ont montré que les étapes précoces de détection sont cruciales pour la pathogénicité des Th17, tandis que l’étape finale de ligature est étonnamment non essentielle. L’ablation de KU80 ou de DNA‑PKcs réduisait nettement la capacité des Th17 à produire des molécules inflammatoires telles que IL‑17A, IL‑2 et GM‑CSF et à provoquer des lésions tissulaires chez la souris. En revanche, la suppression de la ligase IV de l’ADN n’a guère atténué la maladie, bien qu’elle ait perturbé la prolifération cellulaire. Ces expériences indiquent que ce qui compte vraiment pour l’auto‑immunité est l’acte de détection des cassures d’ADN, et pas nécessairement leur réparation effective.
Une enzyme de réparation de l’ADN se comporte comme un amplificateur génique
En approfondissant les mécanismes, l’équipe a découvert que DNA‑PKcs fait plus que rester collée aux extrémités d’ADN brisées. Une fois activée par les capteurs KU, elle se modifie chimiquement au niveau d’une région appelée cluster PQR. Dans cet état, DNA‑PKcs peut se lier physiquement à RORγt, le facteur de transcription maître qui définit les cellules Th17. Ce partenariat stabilise RORγt sur l’ADN à proximité des gènes effecteurs et maintient ces régions de la chromatine ouvertes et accessibles. Lorsque DNA‑PKcs était supprimée, ou lorsque son cluster PQR était spécifiquement éliminé, RORγt ne pouvait plus occuper efficacement les sites clés de gènes inflammatoires, et les Th17 perdaient une grande partie de leur capacité à alimenter l’auto‑immunité. Dans des modèles murins, de telles cellules modifiées ne provoquaient pas d’inflammation rétinienne malgré leur présence dans le système immunitaire. 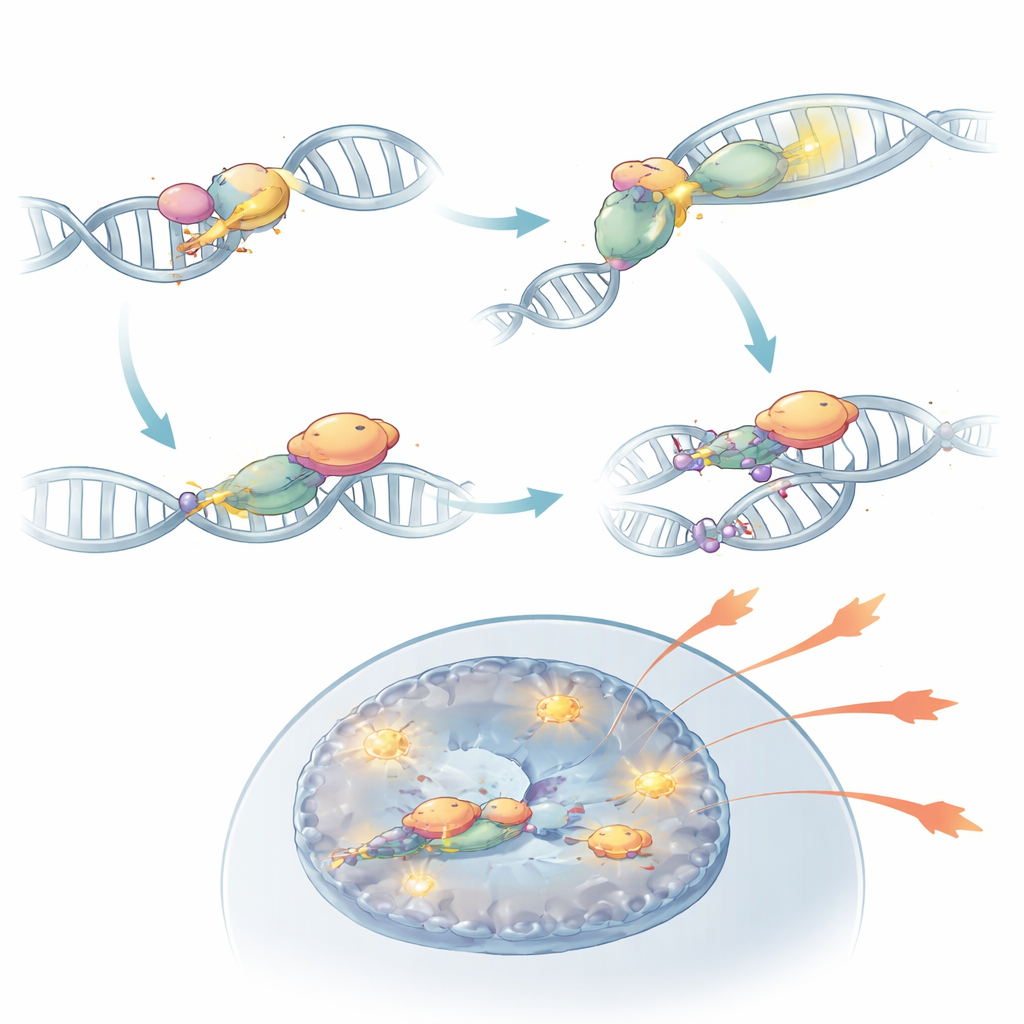
Un signal cytokine qui nourrit réparation et inflammation
L’étude relie également ce réseau de détection de l’ADN à un messager inflammatoire bien connu, la cytokine IL‑23. IL‑23 est reconnue pour pousser les Th17 vers un état néfaste et destructeur pour les tissus. Ici, IL‑23 a été montrée comme augmentant l’activité de la NHEJ, réduisant l’accumulation de cassures d’ADN et renforçant la modification activateur de DNA‑PKcs au site PQR. Cet effet dépendait du récepteur d’IL‑23 : les Th17 dépourvus de ce récepteur ne pouvaient pas activer efficacement la NHEJ et accumulaient davantage de dommages à l’ADN. Par de larges analyses protéiques et transcriptionnelles, les auteurs ont identifié une protéine jusque‑là peu reconnue, IER2, comme un lien entre la signalisation IL‑23 et DNA‑PKcs. IL‑23 induit IER2, qui augmente ensuite l’activité kinase de DNA‑PKcs, soutenant à la fois la réparation de l’ADN et l’expression des gènes inflammatoires pilotée par RORγt.
Un sous‑ensemble Th17 à haut risque chez les patients
Pour relier ces mécanismes à la maladie humaine, les chercheurs ont profilé les cellules immunitaires de patients atteints d’uvéite auto‑immune à différents stades cliniques. Grâce au séquençage ARN unicellulaire, ils ont mis au jour une sous‑population de Th17 présentant de hauts niveaux d’IER2 et de gènes liés à la NHEJ, en particulier des composants de DNA‑PKcs. Ces Th17 à forte expression d’IER2 étaient les plus abondants chez les patients en phase active et récurrente de la maladie et montraient de forts signatures métaboliques, de signalisation et d’inflammation. Ils portaient aussi des marqueurs d’une réparation de l’ADN efficace avec moins de cassures non résolues. L’analyse computationnelle suggérait que, à mesure que les Th17 progressent le long d’une trajectoire développementale vers une plus grande expression d’IER2, leur potentiel inflammatoire augmente parallèlement, faisant de ce sous‑ensemble un probable moteur d’auto‑immunité sévère.
Transformer un système protecteur en cible thérapeutique
Au total, ce travail dresse le portrait d’un système détecteur de réparation de l’ADN — destiné à protéger le génome — qui sert aussi de régulateur des réponses immunitaires dangereuses. Les Th17 pathogéniques exploitent la détection NHEJ, l’activation de DNA‑PKcs et l’axe IL‑23–IER2 pour stabiliser RORγt et maintenir un état inflammatoire à haut niveau tout en maîtrisant les dommages à l’ADN. Pour les patients, cela ouvre de nouvelles pistes thérapeutiques : plutôt que de supprimer globalement le système immunitaire, des médicaments visant spécifiquement l’interférence avec la détection des cassures médiée par KU, l’activation de DNA‑PKcs au niveau du cluster PQR, ou l’interaction IER2–DNA‑PKcs pourraient réduire la pathogénicité des Th17 et calmer l’auto‑immunité sans bloquer complètement la réparation essentielle de l’ADN dans d’autres cellules.
Citation: Chen, GY., Zhu, WJ., Li, Z. et al. Sensing of DNA double-strand breaks by the NHEJ system stabilizes RORγt transcriptional activity and shapes Th17 pathogenicity in autoimmunity. Cell Res 36, 340–358 (2026). https://doi.org/10.1038/s41422-025-01204-6
Mots-clés: Cellules Th17, Réparation de l’ADN, Auto-immunité, DNA-PKcs, Signalisation IL-23