Clear Sky Science · pt
Detecção de quebras de dupla hélice do DNA pelo sistema NHEJ estabiliza a atividade transcricional de RORγt e molda a patogenicidade Th17 na autoimunidade
Quando o Reparo de DNA Transforma Fogo Amigo em Doença
Nosso sistema imune depende de células T para combater infecções, mas às vezes esses defensores se tornam descontrolados e atacam o próprio corpo, causando doenças autoimunes como uveíte no olho ou doença inflamatória intestinal. Este estudo revela uma reviravolta surpreendente: um sistema de reparo de DNA que normalmente protege as células do dano também pode agir como um acelerador oculto para um tipo particularmente agressivo de célula imune, ajudando a impulsionar a inflamação crônica em vez de prevenir o dano.
Dano ao DNA como um Sinal de Alarme Oculto
Quando as células T são fortemente ativadas, especialmente por autoantígenos em contextos autoimunes, suas mitocôndrias produzem rajadas de espécies reativas de oxigênio que podem fragmentar o DNA em quebras de dupla hélice. Normalmente, muitas dessas quebras são letais. Ainda assim, os autores descobriram que um subconjunto chamado células Th17 patogênicas não apenas sobrevive a esse estresse, como o utiliza a seu favor. Essas células exibem altos níveis de uma via de reparo de DNA conhecida como junção de extremidades não homólogas (NHEJ), que detecta rapidamente as quebras e responde. Não se trata apenas de remendar o DNA: a detecção das quebras em si parece alimentar o programa que torna as células Th17 inflamatórias e causadoras de doença, particularmente em modelos murinos de uveíte autoimune e colite. 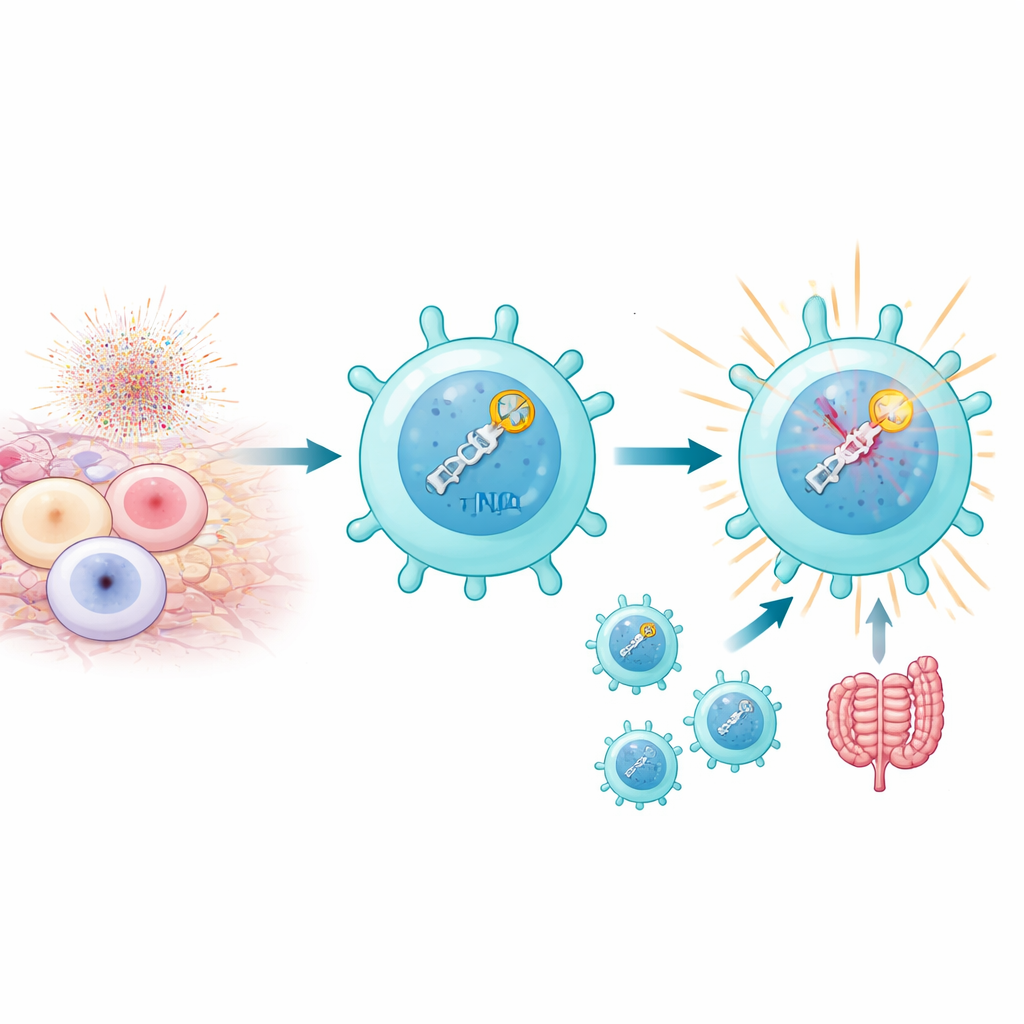
Sensores de Reparo, Não o Reparo em Si, Causam Dano
A NHEJ é um processo em múltiplas etapas, começando com proteínas sensoras (KU70 e KU80) que capturam as extremidades do DNA quebrado e recrutam uma grande enzima, DNA-PKcs, que por sua vez atrai ligases que selam a quebra. Ao desativar seletivamente diferentes partes dessa via em células T, os pesquisadores mostraram que as etapas iniciais de detecção são cruciais para a patogenicidade Th17, enquanto a etapa final de ligação é surpreendentemente dispensável. A eliminação de KU80 ou DNA-PKcs reduziu fortemente a capacidade das células Th17 de produzir moléculas inflamatórias como IL-17A, IL-2 e GM-CSF e de causar dano tecidual em camundongos. Em contraste, remover a ligase IV do DNA pouco atenuou a doença, embora prejudicasse a proliferação celular. Esses experimentos indicam que o que realmente importa para a autoimunidade é o ato de detectar quebras de DNA, não necessariamente repará-las.
Uma Enzima de Reparo do DNA que Atua Também como Estabilizadora de Genes
Aprofundando, a equipe descobriu que DNA-PKcs faz mais do que apenas se posicionar nas extremidades do DNA quebrado. Uma vez ativado pelos sensores KU, ele se modifica quimicamente em uma região chamada cluster PQR. Nesse estado, o DNA-PKcs pode se ligar fisicamente à RORγt, o fator de transcrição mestre que define as células Th17. Essa parceria estabiliza a presença de RORγt no DNA próximo a genes efetores e mantém essas regiões de cromatina abertas e acessíveis. Quando DNA-PKcs foi removido, ou quando seu cluster PQR foi especificamente deletado, RORγt deixou de ocupar efetivamente sítios-chave de genes inflamatórios, e as células Th17 perderam grande parte de seu poder de alimentar a autoimunidade. Em modelos murinos, tais células modificadas não conseguiram inflamar a retina apesar de estarem presentes no sistema imune. 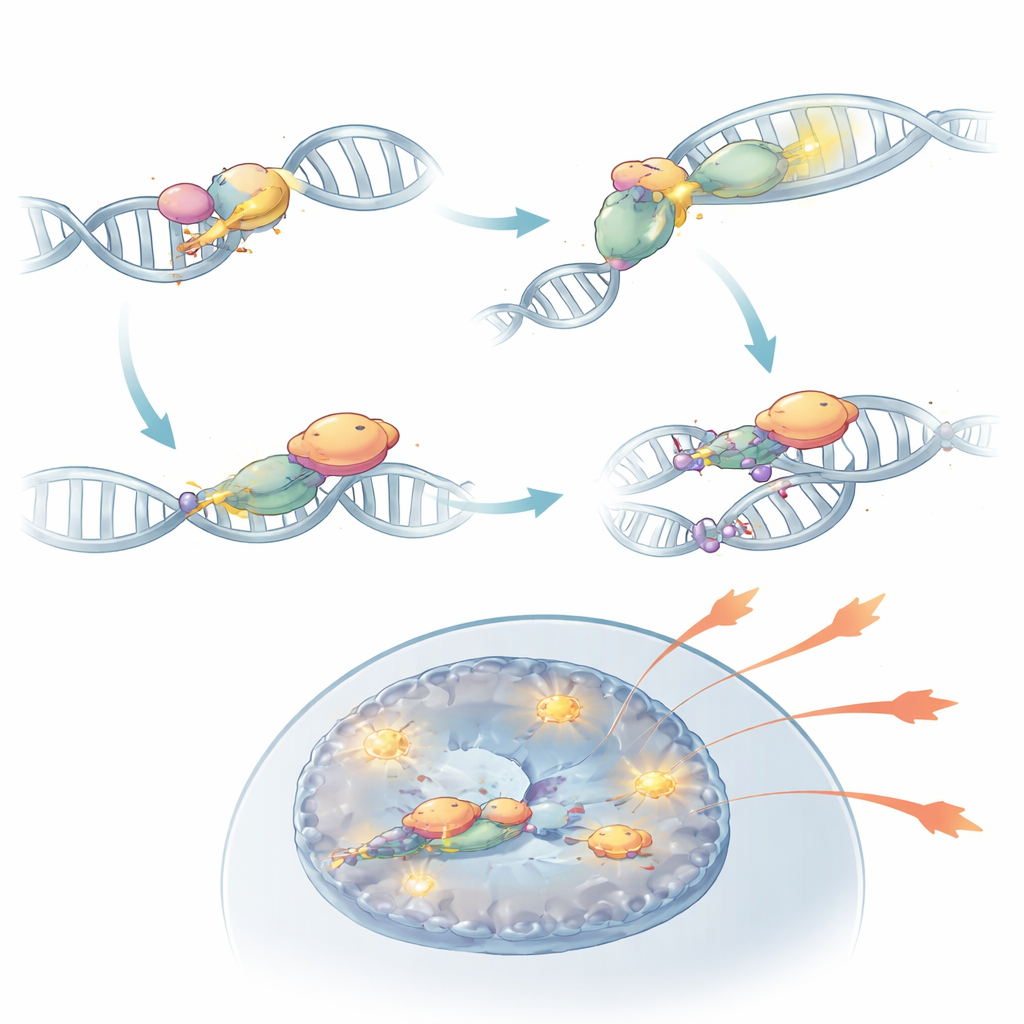
Um Sinal de Citocina que Alimenta Reparo e Inflamação
O estudo também conecta essa rede de detecção de DNA a um mensageiro inflamatório bem conhecido, a citocina IL-23. Sabe-se que IL-23 impulsiona as células Th17 para um estado prejudicial e danificador de tecido. Aqui, mostrou-se que IL-23 aumenta a atividade da NHEJ, reduz o acúmulo de quebras de DNA e potencializa a modificação ativadora de DNA-PKcs no sítio PQR. Esse efeito dependia do receptor de IL-23: células Th17 sem o receptor não conseguiam ativar eficientemente a NHEJ e acumulavam mais danos no DNA. Por meio de análises em larga escala de proteínas e RNA, os autores identificaram uma proteína previamente pouco apreciada, IER2, como uma ponte entre a sinalização de IL-23 e o DNA-PKcs. IL-23 induz IER2, que então potencializa a atividade quinase do DNA-PKcs, sustentando tanto o reparo de DNA quanto a expressão de genes inflamatórios dirigida por RORγt.
Um Subconjunto Th17 de Alto Risco em Pacientes
Para conectar esses mecanismos à doença humana, os pesquisadores perfilaram células imunes de pacientes com uveíte autoimune em diferentes estágios clínicos. Usando sequenciamento de RNA de célula única, descobriram uma subpopulação Th17 com altos níveis de IER2 e genes relacionados à NHEJ, particularmente componentes de DNA-PKcs. Essas células Th17 com IER2 alto foram mais abundantes em pacientes com doença ativa e recorrente e exibiram fortes assinaturas de atividade metabólica, sinalização e inflamação. Também mostraram marcadores de reparo de DNA eficiente e menos quebras não resolvidas. Análises computacionais sugeriram que, à medida que as células Th17 progridem ao longo de uma trajetória de desenvolvimento rumo a maior expressão de IER2, seu potencial inflamatório aumenta em paralelo, marcando esse subconjunto como provável motor de autoimunidade severa.
Transformando um Sistema Protetor em Alvo Terapêutico
No geral, o trabalho traça um quadro em que um sistema sensor de reparo de DNA — destinado a proteger o genoma — funciona também como um botão de controle para respostas imunes perigosas. Células Th17 patogênicas exploram a detecção NHEJ, a ativação de DNA-PKcs e o eixo IL-23–IER2 para estabilizar RORγt e manter um estado inflamatório de alta produção enquanto mantêm o dano ao DNA sob controle. Para pacientes, isso sugere novas abordagens terapêuticas: em vez de suprimir amplamente o sistema imune, fármacos que interfiram seletivamente na detecção de quebras mediada por KU, na ativação de DNA-PKcs no cluster PQR ou na interação IER2–DNA-PKcs poderiam reduzir a patogenicidade Th17 e acalmar a doença autoimune sem bloquear completamente o reparo essencial do DNA em outras células.
Citação: Chen, GY., Zhu, WJ., Li, Z. et al. Sensing of DNA double-strand breaks by the NHEJ system stabilizes RORγt transcriptional activity and shapes Th17 pathogenicity in autoimmunity. Cell Res 36, 340–358 (2026). https://doi.org/10.1038/s41422-025-01204-6
Palavras-chave: células Th17, reparo de DNA, autoimunidade, DNA-PKcs, sinalização IL-23