Clear Sky Science · pt
Um modelo explicável generativo para previsão de peptídeos antimicrobianos usando rede neural convolucional temporal bidirecional
Combater infecções e câncer com peptídeos inteligentes
Medicamentos capazes de matar microrganismos perigosos e células cancerígenas sem prejudicar tecidos saudáveis são um sonho antigo da medicina. A natureza já produz tais moléculas: peptídeos antimicrobianos, pequenos fragmentos proteicos que perfuram membranas microbianas e também podem modular o sistema imune. Mas encontrar os peptídeos mais promissores entre o vasto número de sequências de aminoácidos possíveis é como procurar agulhas em um palheiro. Este estudo apresenta uma poderosa estrutura de inteligência artificial, GAC-BiTCNN-AMP, que aprende a partir de grandes conjuntos de dados biológicos para prever quais peptídeos provavelmente serão agentes antimicrobianos eficazes e potenciais terapêuticos contra o câncer.
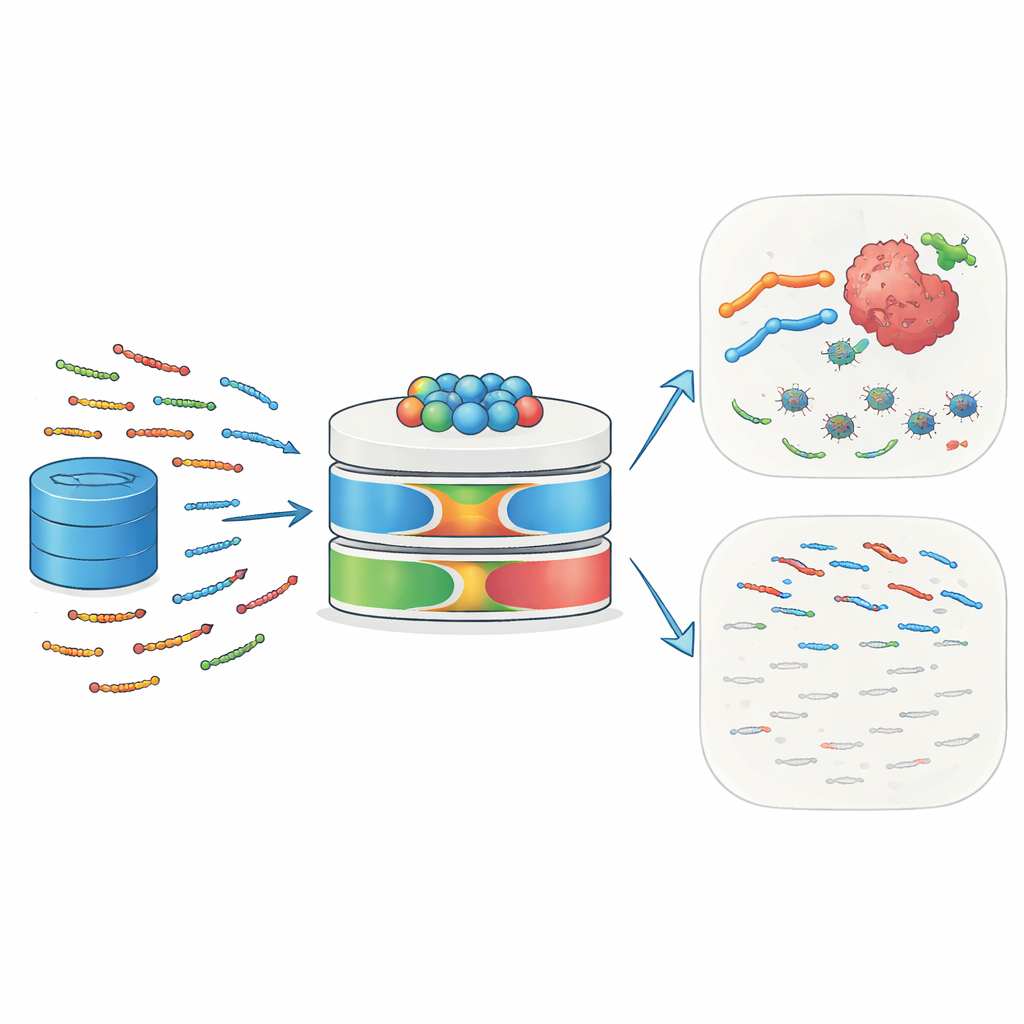
Os pequenos guardiões da natureza
Peptídeos antimicrobianos (AMPs) são cadeias curtas de 10 a 50 aminoácidos encontradas em humanos, animais, plantas e microrganismos. Eles atuam como primeira linha de defesa ao se ligar às superfícies microbianas, perturbar suas membranas e desencadear a morte celular. Muitos AMPs também recrutam células imunes, influenciam inflamações e remodelam o ambiente tecidual local. Células tumorais, com suas membranas incomumente carregadas e desordenadas, podem ser especialmente vulneráveis a esses peptídeos. Alguns exemplos bem conhecidos — incluindo melitina e defensinas — demonstraram capacidade de matar células tumorais, torná-las mais sensíveis à quimioterapia ou radiação e estimular respostas imunes contra tumores. Esse papel duplo contra infecção e câncer torna os AMPs candidatos atraentes para medicamentos de precisão de próxima geração.
Por que as ferramentas tradicionais de predição ficam aquém
Apesar do potencial, identificar novos AMPs in silico continua difícil. Modelos computacionais anteriores baseavam-se principalmente em padrões simples de sequência e em técnicas clássicas de aprendizado de máquina. Frequentemente negligenciavam informações mais ricas sobre como os aminoácidos interagem a longa distância em uma proteína, como essas sequências evoluíram e quais características físicas sutis tornam um peptídeo ao mesmo tempo ativo e seletivo. Muitos modelos usaram dados de treinamento limitados ou redundantes, pularam a seleção sistemática de características e ofereceram pouca interpretabilidade — os pesquisadores não podiam ver facilmente quais aspectos da entrada impulsionavam uma predição. Como resultado, sua precisão e capacidade de generalizar para novos peptídeos eram limitadas, e tinham dificuldade em capturar os diversos papéis biológicos que os AMPs podem desempenhar.
Construindo uma imagem mais rica a partir das sequências
Para superar essas lacunas, os autores primeiro montaram um grande conjunto de dados cuidadosamente filtrado a partir de seis bases de AMPs e da UniProt. Eles distinguiram peptídeos ativos de inativos usando critérios experimentais rígidos e reduziram a redundância para que sequências intimamente relacionadas não inflassem o desempenho. Em seguida, transformaram cada sequência peptídica em múltiplas visões numéricas complementares. Três modelos de linguagem para proteínas de ponta — ProtTrans-T5, UniRep e ESM-2 — foram usados para gerar embeddings de alta dimensão que codificam contexto, dependências de longo alcance e padrões evolutivos aprendidos a partir de milhões de proteínas. Um descritor personalizado chamado PsePSSM-DCT adicionou informação sobre como cada posição na sequência tende a mutar na evolução e como esses padrões variam suavemente ao longo da sequência. Uma etapa de seleção de características baseada em XGBoost então destilou essas representações ricas até os componentes mais informativos, reduzindo o ruído enquanto preservava o sinal.
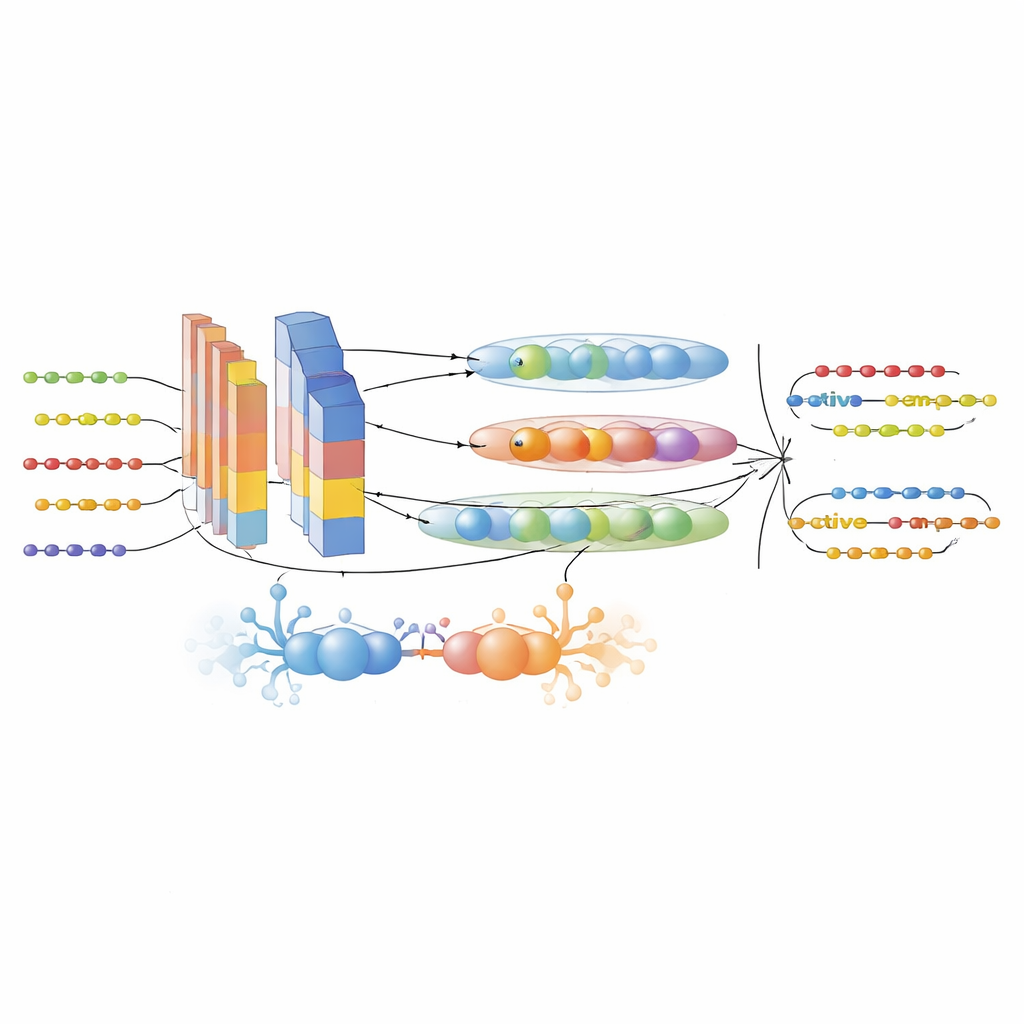
Um motor híbrido de IA para descoberta de peptídeos
No coração da estrutura está o modelo GAC-BiTCNN, uma arquitetura híbrida de aprendizado profundo especificamente projetada para dados de sequência. Ele combina várias ideias: um módulo generativo adversarial que cria vetores de características sintéticas realistas para balancear e enriquecer o conjunto de treinamento; camadas convolucionais que detectam motivos locais; uma rede convolucional temporal bidirecional que captura padrões que correm tanto para frente quanto para trás ao longo da sequência; e redes de cápsulas que agrupam características relacionadas em pequenas “cápsulas” vetoriais, preservando relações hierárquicas. Cada tipo de característica — embeddings de modelos de linguagem e descritores evolutivos — é processado em seu próprio fluxo e posteriormente fundido. O modelo foi treinado e ajustado usando validação cruzada e então testado em um conjunto de dados independente e separado no tempo de entradas de peptídeos mais recentes para minimizar vazamento de informação.
Desempenho, explicabilidade e o que isso significa
GAC-BiTCNN-AMP atingiu desempenho de destaque: até cerca de 97% de acurácia e pontuações de área sob a curva quase perfeitas na validação cruzada, e mais de 95% de acurácia no conjunto de teste independente, superando uma série de preditores AMP existentes e até baselines sintonizados apenas com transformers. Quando os diferentes tipos de características foram combinados, os resultados melhoraram ainda mais, mostrando que cada uma contribui com conhecimento complementar sobre o comportamento peptídico. Para sondar o que o modelo havia aprendido, os autores usaram SHAP, uma técnica popular de IA explicável, para medir como diferentes características latentes influenciavam as predições. Embora essas características sejam abstratas, a análise confirmou que o modelo se apoia em um conjunto compacto de padrões discriminativos biologicamente significativos em vez de ruído aleatório. Em termos simples, o sistema parece estar “observando” os tipos certos de sinais.
O que isso significa para medicamentos futuros
Para não especialistas, a principal conclusão é que este trabalho fornece um filtro orientado por dados e de alta precisão para peneirar vastos números de sequências peptídicas e identificar aquelas com maior probabilidade de agir como agentes antimicrobianos ou anticâncer eficazes. Ao mesclar modelagem generativa, múltiplos modelos de linguagem para proteínas e aprendizado profundo explicável, o GAC-BiTCNN-AMP oferece uma maneira escalável de priorizar candidatos para testes laboratoriais, potencialmente acelerando o desenvolvimento de novos tratamentos para infecções e cânceres que resistem às terapias atuais. Extensões futuras podem não apenas prever quais peptídeos funcionam, mas também orientar o desenho de sequências inteiramente novas ajustadas para potência, seletividade e segurança.
Citação: Ali, F., Khalid, M., Alsini, R. et al. A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network. Sci Rep 16, 13801 (2026). https://doi.org/10.1038/s41598-026-43370-6
Palavras-chave: peptídeos antimicrobianos, modelos de linguagem para proteínas, aprendizado profundo, oncologia de precisão, descoberta de fármacos