Clear Sky Science · pl
Generatywny model wyjaśnialny do przewidywania peptydów przeciwdrobnoustrojowych z użyciem dwukierunkowej splotowej sieci czasowej
Walka z zakażeniami i rakiem przy pomocy inteligentnych peptydów
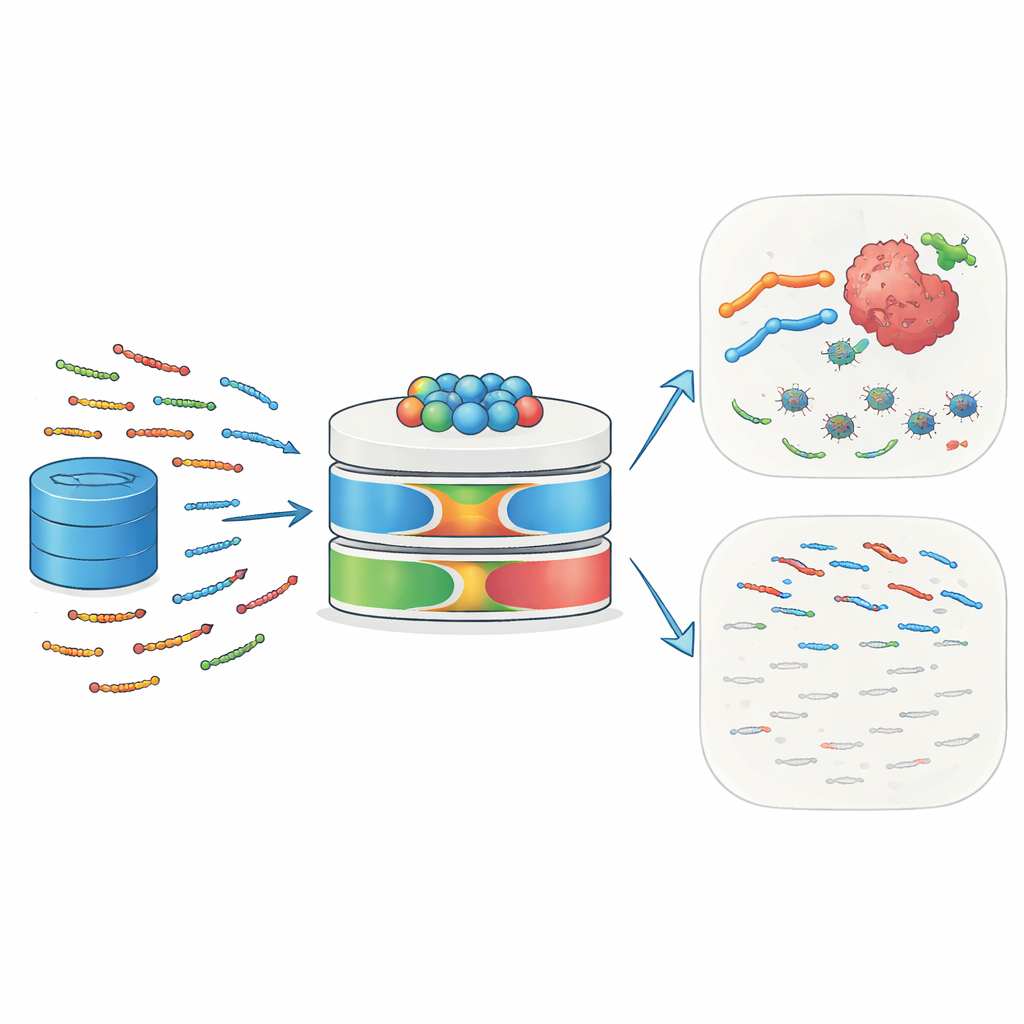
Leki zdolne zabijać niebezpieczne drobnoustroje i komórki nowotworowe bez szkody dla zdrowych tkanek to od dawna aspiracja medycyny. Natura już produkuje takie cząsteczki: peptydy przeciwdrobnoustrojowe, niewielkie fragmenty białek, które perforują błony mikroorganizmów i potrafią też modyfikować układ odpornościowy. Jednak znalezienie najbardziej obiecujących peptydów spośród ogromnej liczby możliwych sekwencji aminokwasów przypomina szukanie igieł w stogu siana. W tym badaniu zaproponowano wydajne ramy sztucznej inteligencji, GAC-BiTCNN-AMP, które uczą się na dużych zbiorach biologicznych, aby przewidywać, które peptydy mają największe szanse być skutecznymi środkami przeciwdrobnoustrojowymi oraz potencjalnymi terapiami przeciwnowotworowymi.
Małe straże natury
Peptydy przeciwdrobnoustrojowe (AMP) to krótkie łańcuchy 10–50 aminokwasów występujące u ludzi, zwierząt, roślin i mikroorganizmów. Działają jako pierwsza linia obrony, wiążąc się z powierzchniami mikroorganizmów, zaburzając ich błony i wywołując śmierć komórki. Wiele AMP również rekrutuje komórki odpornościowe, wpływa na stany zapalne i przekształca lokalne środowisko tkankowe. Komórki nowotworowe, ze swoimi nietypowo naładowanymi i zaburzonymi błonami, mogą być szczególnie podatne na takie peptydy. Niektóre znane przykłady — w tym melittina i defensyny — wykazały zdolność do zabijania komórek nowotworowych, uczulania ich na chemioterapię lub radioterapię oraz stymulowania odpowiedzi immunologicznych przeciw nowotworom. Ta podwójna rola wobec zakażeń i raka czyni AMP atrakcyjnymi kandydatami do leków następnej generacji.
Dlaczego tradycyjne narzędzia zawodzą
Mimo obietnic, identyfikacja nowych AMP in silico pozostaje trudna. Wcześniejsze modele komputerowe opierały się głównie na prostych wzorcach sekwencji i klasycznych technikach uczenia maszynowego. Często pomijały bogatsze informacje o tym, jak aminokwasy oddziałują na długich dystansach w białku, jak sekwencje ewoluowały oraz które subtelne cechy fizyczne czynią peptyd aktywnym i selektywnym. Wiele modeli korzystało z ograniczonych lub redundantnych danych treningowych, pomijało systematyczny wybór cech i oferowało niewielką interpretowalność — badacze nie mogli łatwo zobaczyć, które aspekty wejścia decydowały o predykcji. W efekcie ich dokładność i zdolność uogólniania na nowe peptydy były ograniczone, a modele miały trudności z uchwyceniem zróżnicowanych biologicznych ról AMP.
Budowanie pełniejszego obrazu z sekwencji
Aby wypełnić te luki, autorzy najpierw zgromadzili duży, starannie filtrowany zbiór danych z sześciu baz AMP i UniProt. Rozróżnili peptydy aktywne od nieaktywnych przy użyciu rygorystycznych kryteriów eksperymentalnych i zredukowali redundancję, aby blisko spokrewnione sekwencje nie zawyżały wyników. Następnie przekształcili każdą sekwencję peptydu w wiele komplementarnych widoków numerycznych. Trzy nowoczesne modele języka białek — ProtTrans-T5, UniRep i ESM-2 — zostały użyte do wygenerowania wysokowymiarowych osadzeń kodujących kontekst, zależności długodystansowe i wzorce ewolucyjne wyuczone na milionach białek. Własny deskryptor nazwany PsePSSM-DCT dodał informacje o tym, jak każda pozycja w sekwencji ma tendencję do mutowania w ewolucji i jak te wzorce zmieniają się płynnie wzdłuż sekwencji. Etap selekcji cech oparty na XGBoost następnie wydestylował te bogate reprezentacje do najbardziej informacyjnych komponentów, odcinając szum przy zachowaniu sygnału.
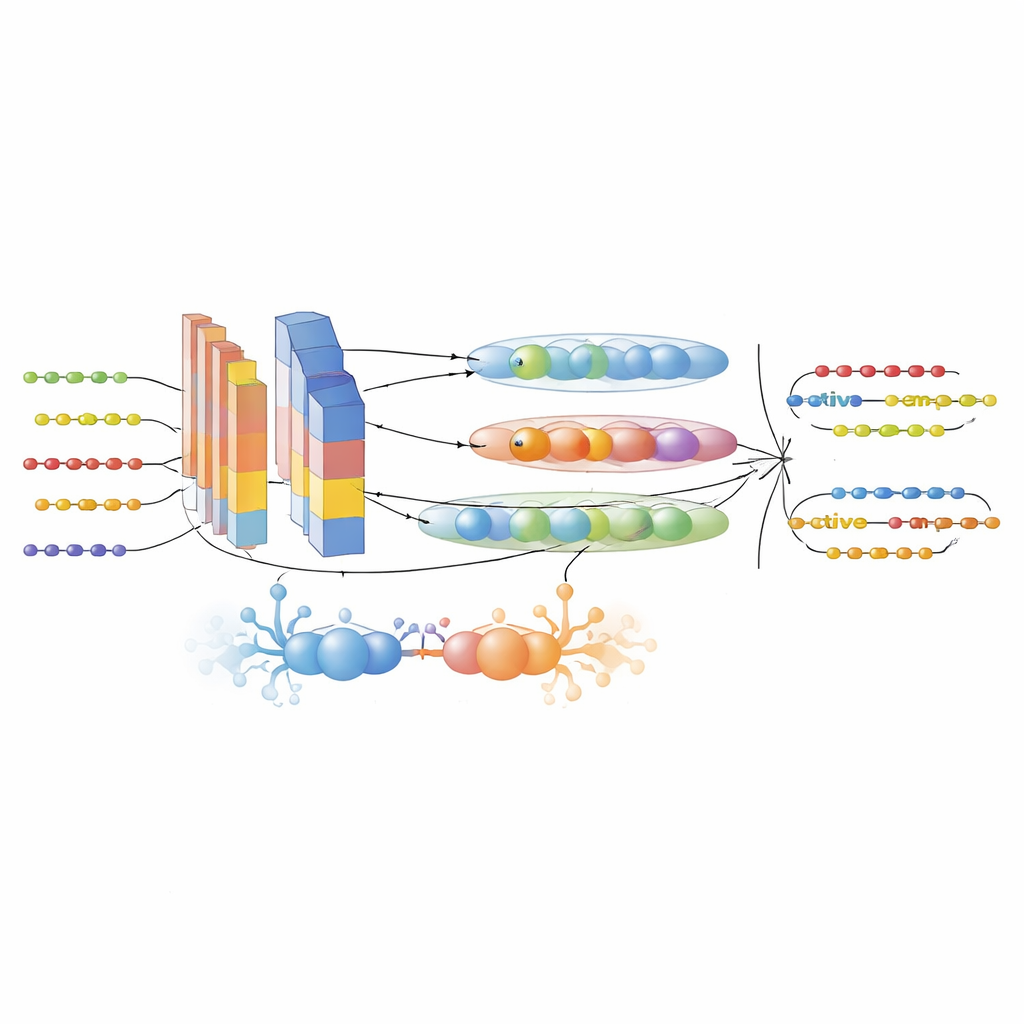
Hybrydowy silnik AI do odkrywania peptydów
Rdzeniem ram jest model GAC-BiTCNN, hybrydowa architektura głębokiego uczenia specjalnie dostosowana do danych sekwencyjnych. Łączy kilka pomysłów: moduł generatywno-antagonistyczny, który tworzy realistyczne syntetyczne wektory cech, by zrównoważyć i wzbogacić zbiór treningowy; warstwy splotowe wykrywające lokalne motywy; dwukierunkową splotową sieć czasową chwytającą wzorce biegnące zarówno w przód, jak i w tył wzdłuż sekwencji; oraz sieci kapsułkowe grupujące powiązane cechy w małe wektorowe „kapsuły”, zachowując relacje hierarchiczne. Każdy typ cech — osadzenia modeli języka białek i deskryptory ewolucyjne — jest przetwarzany w osobnym strumieniu i później łączony. Model był trenowany i strojony z użyciem walidacji krzyżowej, a następnie testowany na całkowicie odrębnym, czasowo oddzielonym zbiorze nowszych wpisów peptydów, by zminimalizować wyciek informacji.
Wydajność, wyjaśnialność i ich znaczenie
GAC-BiTCNN-AMP osiągnął znakomite wyniki: do około 97% dokładności i niemal doskonałe wyniki AUC w walidacji krzyżowej oraz ponad 95% dokładności na niezależnym zbiorze testowym, przewyższając szereg istniejących predyktorów AMP, a nawet bazowe modele typu transformer z fine-tuningiem. Gdy różne typy cech zostały połączone, wyniki jeszcze się poprawiły, pokazując, że każda dostarcza komplementarnej wiedzy o zachowaniu peptydu. Aby zbadać, czego model się nauczył, autorzy zastosowali SHAP, popularną technikę wyjaśnialnej AI, aby zmierzyć, jak różne ukryte cechy wpływają na predykcje. Chociaż te cechy są abstrakcyjne, analiza potwierdziła, że model opiera się na zwartej grupie dyskryminujących, biologicznie sensownych wzorców, a nie na losowym szumie. Mówiąc wprost, system zdaje się „patrzeć” na właściwe rodzaje sygnałów.
Co to oznacza dla przyszłych leków
Dla osób niebędących specjalistami kluczowy wniosek jest taki, że praca ta dostarcza wysoce dokładnego, opartego na danych filtra do przesiania ogromnych liczb sekwencji peptydowych w celu wytypowania tych, które najprawdopodobniej będą działać jako skuteczne środki przeciwdrobnoustrojowe lub przeciwnowotworowe. Poprzez połączenie modelowania generatywnego, wielu modeli języka białek i wyjaśnialnego uczenia głębokiego, GAC-BiTCNN-AMP oferuje skalowalny sposób priorytetyzacji kandydatów do testów laboratoryjnych, potencjalnie przyspieszając rozwój nowych terapii przeciw infekcjom i nowotworom opornym na obecne terapie. Przyszłe rozszerzenia mogą nie tylko przewidywać, które peptydy działają, lecz także kierować projektowaniem całkowicie nowych sekwencji dobranych pod kątem mocy, selektywności i bezpieczeństwa.
Cytowanie: Ali, F., Khalid, M., Alsini, R. et al. A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network. Sci Rep 16, 13801 (2026). https://doi.org/10.1038/s41598-026-43370-6
Słowa kluczowe: peptydy przeciwdrobnoustrojowe, modele języka białek, uczenie głębokie, onkologia precyzyjna, odkrywanie leków