Clear Sky Science · en
A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network
Fighting infections and cancer with smart peptides
Drugs that can kill dangerous microbes and cancer cells without harming healthy tissue are a long-standing dream in medicine. Nature already makes such molecules: antimicrobial peptides, tiny protein fragments that punch holes in microbial membranes and can also modulate the immune system. But finding the most promising peptides among the vast number of possible amino acid sequences is like searching for needles in a haystack. This study introduces a powerful artificial intelligence framework, GAC-BiTCNN-AMP, that learns from large biological datasets to predict which peptides are likely to be effective antimicrobial agents and potential cancer therapeutics.
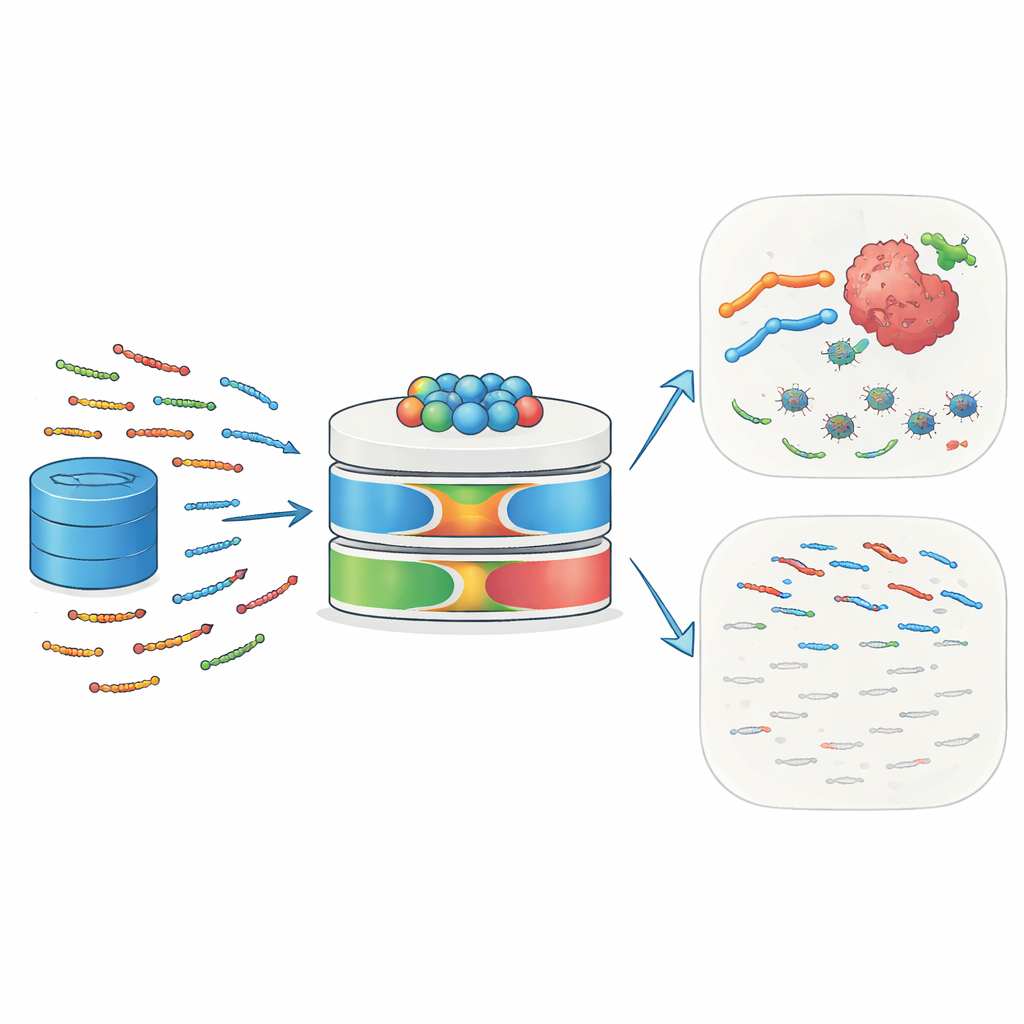
Nature’s tiny bodyguards
Antimicrobial peptides (AMPs) are short chains of 10 to 50 amino acids found in humans, animals, plants, and microbes. They act as a first line of defense by binding to microbial surfaces, disturbing their membranes, and triggering cell death. Many AMPs also recruit immune cells, influence inflammation, and reshape the local tissue environment. Cancer cells, with their unusually charged and disordered membranes, can be especially vulnerable to such peptides. Some well-known examples—including melittin and defensins—have shown the ability to kill tumor cells, sensitize them to chemotherapy or radiation, and stimulate immune responses against tumors. This dual role against infection and cancer makes AMPs attractive candidates for next-generation precision medicines.
Why traditional prediction tools fall short
Despite their promise, identifying new AMPs in silico remains difficult. Earlier computer models relied mainly on simple sequence patterns and classical machine-learning techniques. They often ignored richer information about how amino acids interact over long distances in a protein, how these sequences evolved, and which subtle physical features make a peptide both active and selective. Many models used limited or redundant training data, skipped systematic feature selection, and offered little interpretability—researchers could not easily see which aspects of the input drove a prediction. As a result, their accuracy and ability to generalize to new peptides were constrained, and they struggled to capture the diverse biological roles AMPs can play.
Building a richer picture from sequences
To overcome these gaps, the authors first assembled a large, carefully filtered dataset from six AMP databases and UniProt. They distinguished active peptides from inactive ones using stringent experimental criteria and reduced redundancy so that closely related sequences would not inflate performance. Next, they transformed each peptide sequence into multiple complementary numerical views. Three state-of-the-art protein language models—ProtTrans-T5, UniRep, and ESM-2—were used to generate high-dimensional embeddings that encode context, long-range dependencies, and evolutionary patterns learned from millions of proteins. A custom descriptor called PsePSSM-DCT added information about how each position in a sequence tends to mutate in evolution and how those patterns vary smoothly along the sequence. A feature-selection step based on XGBoost then distilled these rich representations down to the most informative components, trimming noise while preserving signal.
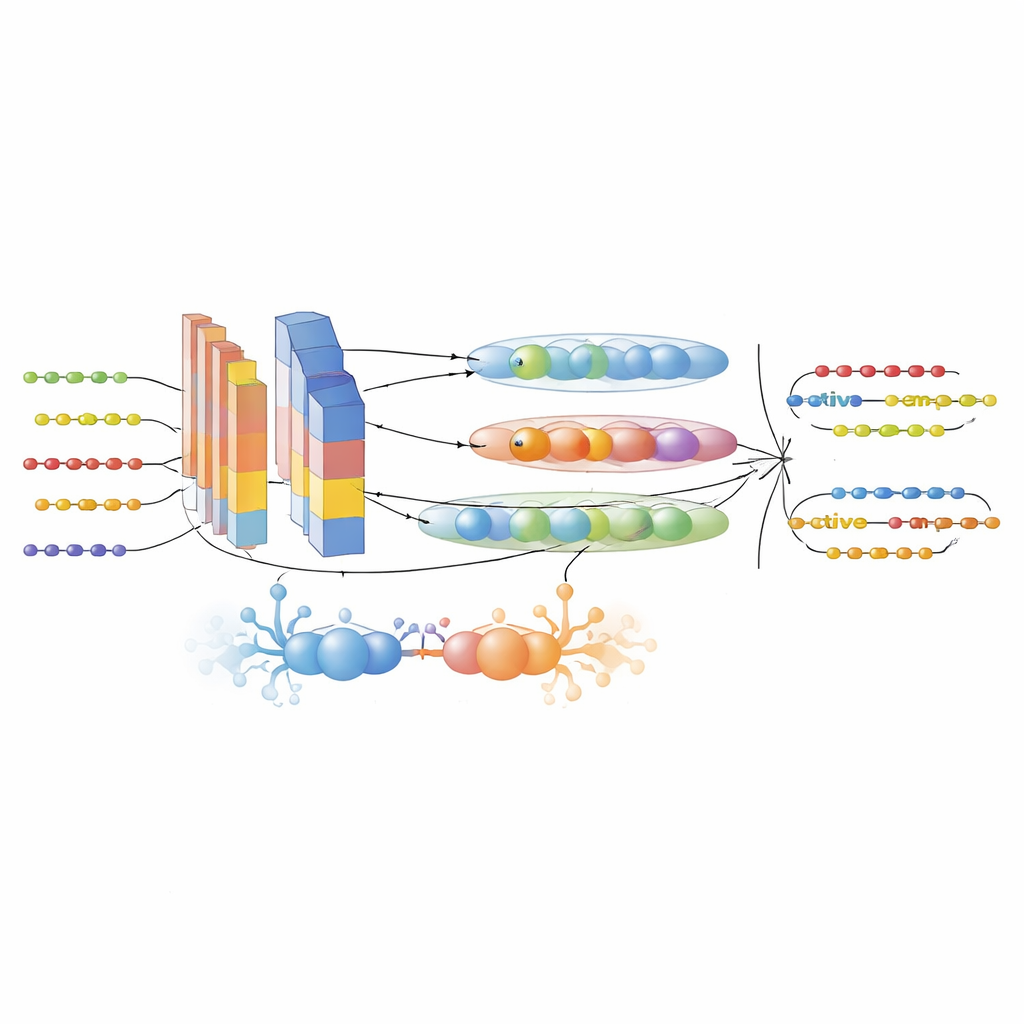
A hybrid AI engine for peptide discovery
The heart of the framework is the GAC-BiTCNN model, a hybrid deep-learning architecture specifically tailored for sequence data. It combines several ideas: a generative adversarial module that creates realistic synthetic feature vectors to balance and enrich the training set; convolutional layers that detect local motifs; a bidirectional temporal convolutional network that captures patterns running both forward and backward along the sequence; and capsule networks that group related features into small vector “capsules,” preserving hierarchical relationships. Each type of feature—language-model embeddings and evolutionary descriptors—is processed in its own stream and later fused. The model was trained and tuned using cross-validation and then tested on a completely separate, time-separated dataset of newer peptide entries to minimize information leakage.
Performance, explainability, and what it means
GAC-BiTCNN-AMP achieved standout performance: up to about 97% accuracy and near-perfect area-under-the-curve scores in cross-validation, and over 95% accuracy on the independent test set, outperforming a range of existing AMP predictors and even fine-tuned transformer-only baselines. When the different feature types were combined, results improved further, showing that each contributes complementary knowledge about peptide behavior. To probe what the model had learned, the authors used SHAP, a popular explainable-AI technique, to measure how different latent features influenced predictions. While these features are abstract, the analysis confirmed that the model relies on a compact set of discriminative, biologically meaningful patterns instead of random noise. In plain terms, the system appears to be “looking” at the right kinds of signals.
What this means for future medicines
For non-specialists, the key takeaway is that this work provides a highly accurate, data-driven filter for sifting through vast numbers of peptide sequences to pinpoint those most likely to act as effective antimicrobial or anti-cancer agents. By blending generative modeling, multiple protein language models, and explainable deep learning, GAC-BiTCNN-AMP offers a scalable way to prioritize candidates for laboratory testing, potentially speeding up the development of new treatments for infections and cancers that resist current therapies. Future extensions may not only predict which peptides work, but also guide the design of entirely new sequences tuned for potency, selectivity, and safety.
Citation: Ali, F., Khalid, M., Alsini, R. et al. A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network. Sci Rep 16, 13801 (2026). https://doi.org/10.1038/s41598-026-43370-6
Keywords: antimicrobial peptides, protein language models, deep learning, precision oncology, drug discovery