Clear Sky Science · it
Un modello generativo spiegabile per la predizione di peptidi antimicrobici usando una rete neurale convoluzionale temporale bidirezionale
Combattere infezioni e cancro con peptidi intelligenti
I farmaci capaci di uccidere microrganismi pericolosi e cellule tumorali senza danneggiare i tessuti sani sono un sogno di lunga data in medicina. La natura produce già queste molecole: i peptidi antimicrobici, frammenti proteici piccoli che perforano le membrane microbiche e possono anche modulare il sistema immunitario. Ma trovare i peptidi più promettenti tra l’enorme numero di possibili sequenze aminoacidiche è come cercare aghi in un pagliaio. Questo studio presenta un potente framework di intelligenza artificiale, GAC-BiTCNN-AMP, che apprende da grandi dataset biologici per prevedere quali peptidi sono probabilmente efficaci come agenti antimicrobici e potenziali terapeutici antitumorali.
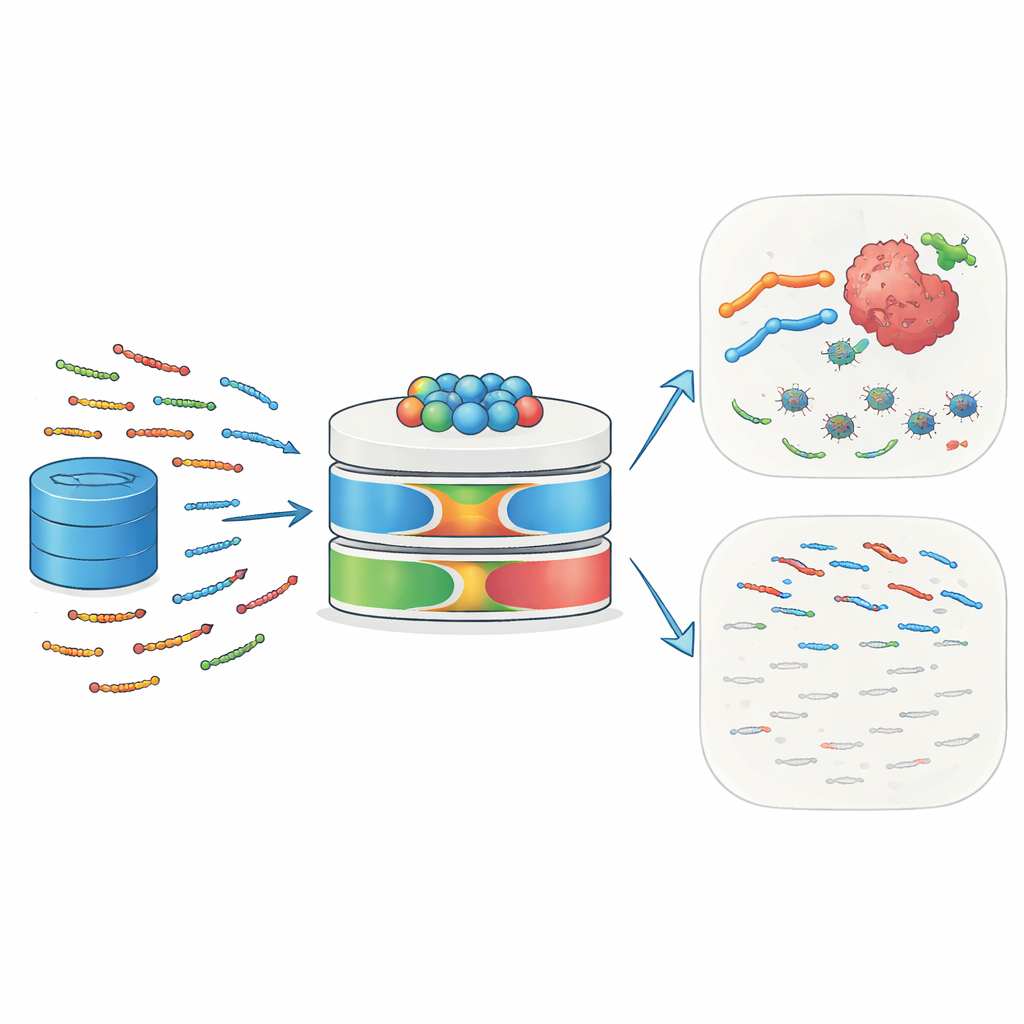
I piccoli guardiani della natura
I peptidi antimicrobici (AMP) sono catene corte di 10–50 amminoacidi presenti in esseri umani, animali, piante e microrganismi. Agiscono come prima linea di difesa legandosi alle superfici microbiche, disturbando le membrane e causando la morte cellulare. Molti AMP reclutano anche cellule immunitarie, influenzano l’infiammazione e rimodellano l’ambiente tissutale locale. Le cellule tumorali, con membrane spesso caricate in modo anomalo e disordinate, possono essere particolarmente vulnerabili a questi peptidi. Alcuni esempi noti — tra cui la melittina e le defensine — hanno mostrato la capacità di uccidere cellule tumorali, sensibilizzarle a chemioterapia o radioterapia e stimolare risposte immunitarie contro i tumori. Questo ruolo duale contro infezioni e cancro rende gli AMP candidati interessanti per le medicine di nuova generazione e di precisione.
Perché gli strumenti tradizionali di predizione non bastano
Nonostante il loro potenziale, identificare nuovi AMP in silico resta difficile. I modelli computazionali precedenti si basavano principalmente su pattern di sequenza semplici e tecniche di machine learning classiche. Spesso ignoravano informazioni più ricche su come gli amminoacidi interagiscono a lunga distanza in una proteina, come queste sequenze sono evolute e quali caratteristiche fisiche sottili rendono un peptide sia attivo sia selettivo. Molti modelli usavano dati di addestramento limitati o ridondanti, trascuravano una selezione sistematica delle caratteristiche e offrivano scarsa interpretabilità — i ricercatori non potevano vedere facilmente quali aspetti dell’input determinavano una predizione. Di conseguenza, accuratezza e capacità di generalizzare a nuovi peptidi erano limitate, e faticavano a cogliere i ruoli biologici diversificati che gli AMP possono svolgere.
Costruire un quadro più ricco a partire dalle sequenze
Per colmare queste lacune, gli autori hanno innanzitutto assemblato un dataset ampio e accuratamente filtrato da sei database di AMP e da UniProt. Hanno distinto i peptidi attivi da quelli inattivi usando criteri sperimentali stringenti e ridotto la ridondanza in modo che sequenze strettamente correlate non gonfiassero le prestazioni. Successivamente, hanno trasformato ogni sequenza peptidica in molteplici viste numeriche complementari. Tre modelli linguistici all’avanguardia per proteine — ProtTrans-T5, UniRep ed ESM-2 — sono stati utilizzati per generare embedding ad alta dimensione che codificano contesto, dipendenze a lungo raggio e pattern evolutivi appresi da milioni di proteine. Un descrittore personalizzato chiamato PsePSSM-DCT ha aggiunto informazioni su come ogni posizione nella sequenza tende a mutare nell’evoluzione e su come tali pattern variano in modo continuo lungo la sequenza. Un passaggio di selezione delle caratteristiche basato su XGBoost ha poi distillato queste rappresentazioni ricche fino ai componenti più informativi, riducendo il rumore e preservando il segnale.
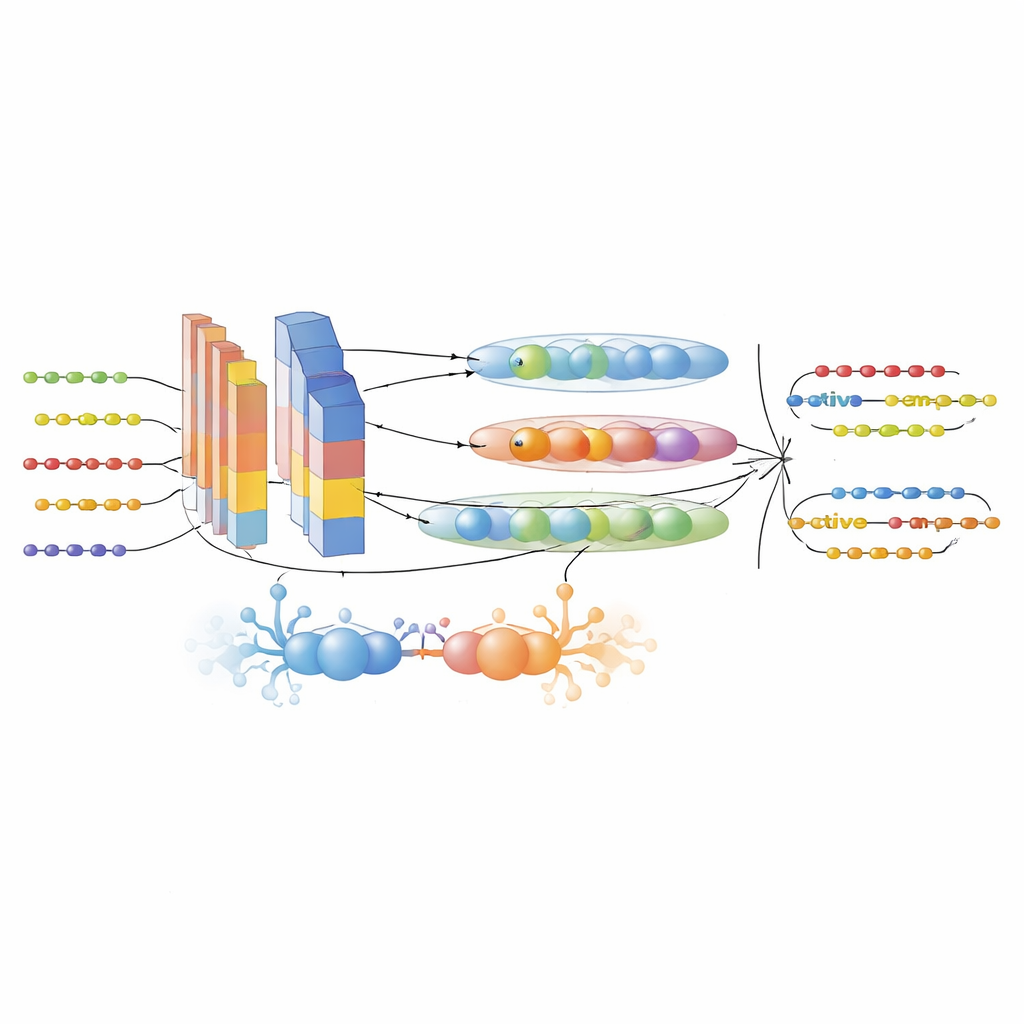
Un motore AI ibrido per la scoperta di peptidi
Il cuore del framework è il modello GAC-BiTCNN, un’architettura di deep learning ibrida specificamente pensata per dati di sequenza. Combina diverse idee: un modulo generativo avversario che crea vettori di caratteristiche sintetici realistici per bilanciare e arricchire il set di addestramento; strati convoluzionali che rilevano motivi locali; una rete convoluzionale temporale bidirezionale che cattura pattern che scorrono sia in avanti sia all’indietro lungo la sequenza; e reti a capsule che raggruppano caratteristiche correlate in piccole “capsule” vettoriali, preservando relazioni gerarchiche. Ogni tipo di caratteristica — embedding dei modelli linguistici ed descrittori evolutivi — è elaborato in un flusso dedicato e poi fuso. Il modello è stato addestrato e ottimizzato usando cross-validation e poi testato su un dataset completamente separato e separato temporalmente, costituito da voci di peptidi più recenti, per minimizzare la fuga di informazioni.
Prestazioni, spiegabilità e implicazioni
GAC-BiTCNN-AMP ha raggiunto performance di rilievo: fino a circa il 97% di accuratezza e punteggi area-under-the-curve quasi perfetti in cross-validation, e oltre il 95% di accuratezza sul set di test indipendente, superando una serie di predittori AMP esistenti e persino baselines basate solo su transformer messi a punto. Quando i diversi tipi di caratteristiche sono stati combinati, i risultati sono ulteriormente migliorati, dimostrando che ciascuno contribuisce con conoscenze complementari sul comportamento dei peptidi. Per indagare ciò che il modello aveva appreso, gli autori hanno usato SHAP, una tecnica di explainable AI popolare, per misurare come diverse caratteristiche latenti influenzassero le predizioni. Pur essendo astratte, le analisi hanno confermato che il modello si basa su un insieme compatto di pattern discriminativi e biologicamente significativi piuttosto che su rumore casuale. In termini semplici, il sistema sembra “guardare” i tipi di segnali giusti.
Cosa significa per le medicine del futuro
Per i non specialisti, il messaggio principale è che questo lavoro fornisce un filtro guidato dai dati, altamente accurato, per setacciare vasti numeri di sequenze peptidiche e individuare quelle più probabilmente efficaci come agenti antimicrobici o antitumorali. Mescolando modellazione generativa, più modelli linguistici per proteine e deep learning spiegabile, GAC-BiTCNN-AMP offre un modo scalabile per dare priorità ai candidati da testare in laboratorio, accelerando potenzialmente lo sviluppo di nuovi trattamenti per infezioni e tumori resistenti alle terapie attuali. Estensioni future potrebbero non solo prevedere quali peptidi funzionano, ma anche guidare la progettazione di sequenze completamente nuove ottimizzate per potenza, selettività e sicurezza.
Citazione: Ali, F., Khalid, M., Alsini, R. et al. A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network. Sci Rep 16, 13801 (2026). https://doi.org/10.1038/s41598-026-43370-6
Parole chiave: peptidi antimicrobici, modelli linguistici per proteine, deep learning, oncologia di precisione, scoperta di farmaci