Clear Sky Science · es
Un modelo generativo explicable para la predicción de péptidos antimicrobianos usando redes neuronales convolucionales temporales bidireccionales
Combatir infecciones y cáncer con péptidos inteligentes
Los fármacos capaces de eliminar microbios peligrosos y células cancerosas sin dañar el tejido sano han sido durante mucho tiempo un sueño en medicina. La naturaleza ya produce tales moléculas: los péptidos antimicrobianos, fragmentos proteicos diminutos que perforan membranas microbianas y también pueden modular el sistema inmune. Pero encontrar los péptidos más prometedores entre la inmensa cantidad de secuencias posibles de aminoácidos es como buscar agujas en un pajar. Este estudio presenta un potente marco de inteligencia artificial, GAC-BiTCNN-AMP, que aprende a partir de grandes conjuntos de datos biológicos para predecir qué péptidos probablemente serán agentes antimicrobianos eficaces y posibles terapias contra el cáncer.
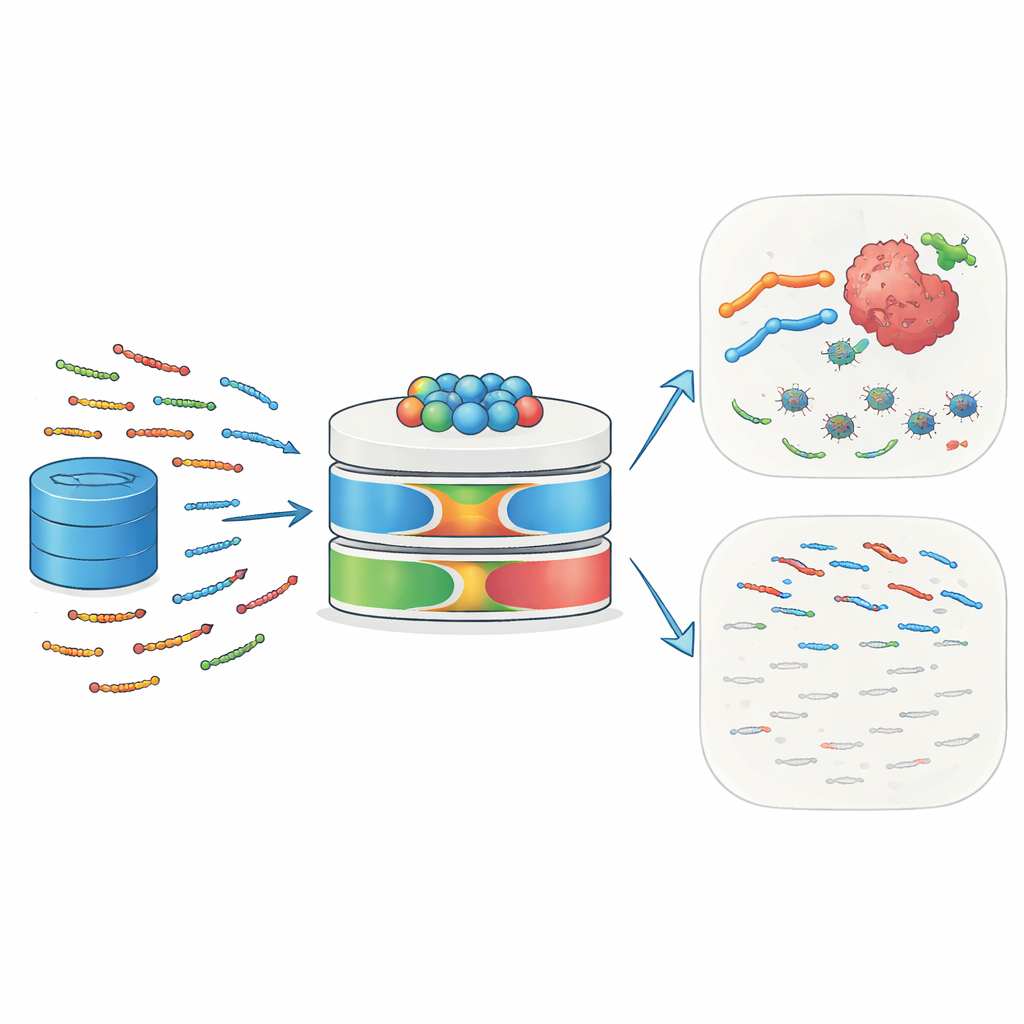
Los pequeños guardianes de la naturaleza
Los péptidos antimicrobianos (AMP) son cadenas cortas de 10 a 50 aminoácidos presentes en humanos, animales, plantas y microbios. Actúan como primera línea de defensa al unirse a las superficies microbianas, alterar sus membranas y desencadenar la muerte celular. Muchos AMP también reclutan células inmunitarias, influyen en la inflamación y remodelan el entorno tisular local. Las células cancerosas, con sus membranas inusualmente cargadas y desordenadas, pueden ser especialmente vulnerables a esos péptidos. Algunos ejemplos conocidos—como la melitina y las defensinas—han demostrado capacidad para matar células tumorales, sensibilizarlas a la quimioterapia o la radiación y estimular respuestas inmunitarias contra los tumores. Este papel dual contra la infección y el cáncer convierte a los AMP en candidatos atractivos para la siguiente generación de medicinas de precisión.
Por qué las herramientas tradicionales de predicción se quedan cortas
A pesar de su potencial, identificar nuevos AMP in silico sigue siendo difícil. Los modelos computacionales previos se basaban principalmente en patrones de secuencia sencillos y técnicas clásicas de aprendizaje automático. A menudo ignoraban información más rica sobre cómo interactúan los aminoácidos a larga distancia en una proteína, cómo evolucionaron estas secuencias y qué características físicas sutiles hacen que un péptido sea tanto activo como selectivo. Muchos modelos usaron datos de entrenamiento limitados o redundantes, omitieron una selección sistemática de características y ofrecieron poca interpretabilidad—los investigadores no podían ver fácilmente qué aspectos de la entrada impulsaban una predicción. Como resultado, su precisión y capacidad de generalizar a nuevos péptidos estaban limitadas, y tenían dificultades para capturar los diversos roles biológicos que los AMP pueden desempeñar.
Construir una imagen más rica a partir de secuencias
Para salvar estas lagunas, los autores primero reunieron un conjunto de datos grande y cuidadosamente filtrado a partir de seis bases de datos de AMP y UniProt. Distinguen péptidos activos de inactivos usando criterios experimentales estrictos y redujeron la redundancia para que secuencias estrechamente relacionadas no inflaran el rendimiento. A continuación, transformaron cada secuencia de péptido en múltiples vistas numéricas complementarias. Tres modelos de lenguaje de proteínas de última generación—ProtTrans-T5, UniRep y ESM-2—se emplearon para generar embeddings de alta dimensión que codifican contexto, dependencias a larga distancia y patrones evolutivos aprendidos a partir de millones de proteínas. Un descriptor personalizado llamado PsePSSM-DCT añadió información sobre cómo tiende a mutar cada posición en una secuencia durante la evolución y cómo esos patrones varían suavemente a lo largo de la secuencia. Un paso de selección de características basado en XGBoost destiló estas representaciones ricas hasta los componentes más informativos, recortando ruido sin perder señal.
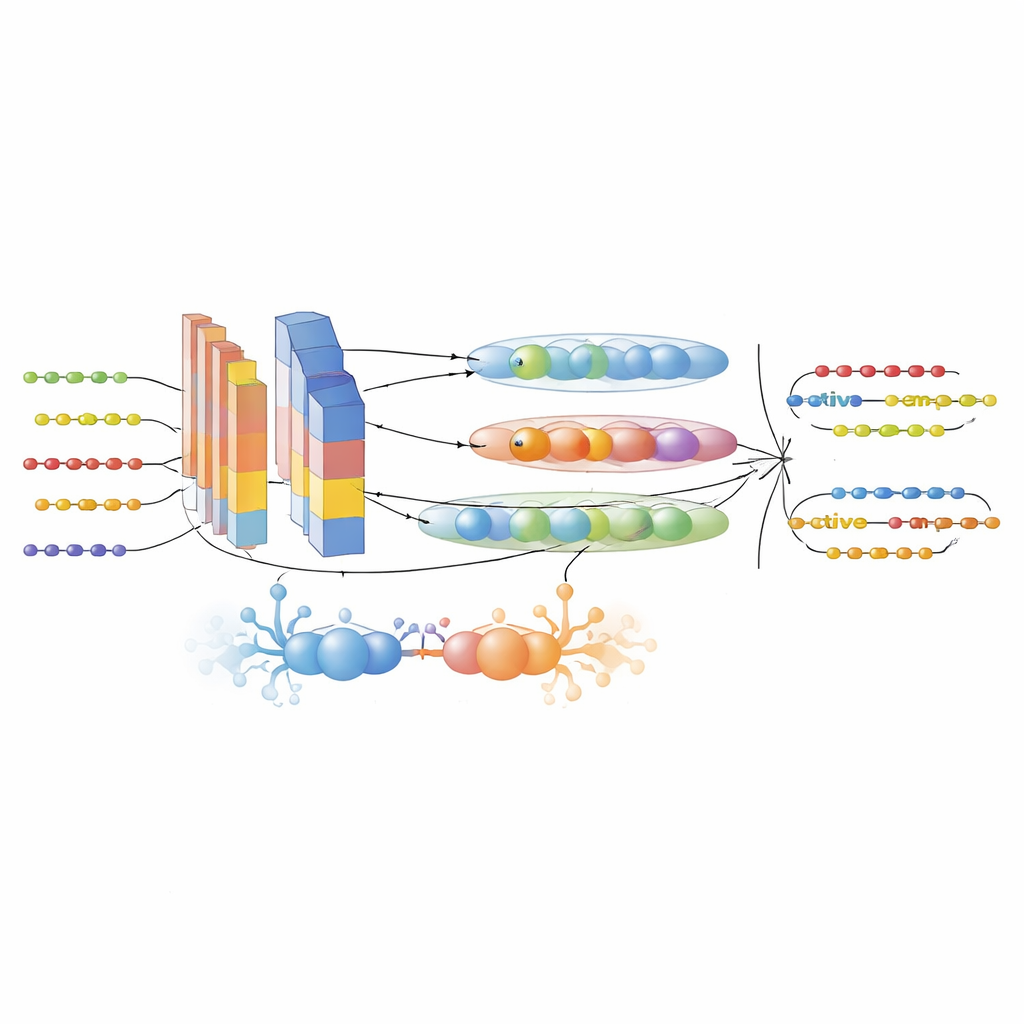
Un motor híbrido de IA para el descubrimiento de péptidos
El núcleo del marco es el modelo GAC-BiTCNN, una arquitectura híbrida de aprendizaje profundo específica para datos de secuencia. Combina varias ideas: un módulo generativo adversarial que crea vectores de características sintéticos realistas para balancear y enriquecer el conjunto de entrenamiento; capas convolucionales que detectan motivos locales; una red convolucional temporal bidireccional que capta patrones que corren tanto hacia adelante como hacia atrás a lo largo de la secuencia; y redes de cápsulas que agrupan características relacionadas en pequeñas “cápsulas” vectoriales, preservando relaciones jerárquicas. Cada tipo de característica—embeddings de modelos del lenguaje y descriptores evolutivos—se procesa en su propio flujo y luego se fusiona. El modelo se entrenó y ajustó mediante validación cruzada y después se probó en un conjunto de datos independiente y separado en el tiempo con entradas de péptidos más recientes para minimizar la filtración de información.
Rendimiento, explicabilidad y su significado
GAC-BiTCNN-AMP alcanzó un rendimiento destacado: hasta alrededor del 97% de precisión y puntuaciones prácticamente perfectas del área bajo la curva en validación cruzada, y más del 95% de precisión en el conjunto de prueba independiente, superando a diversos predictores de AMP existentes e incluso a líneas base basadas únicamente en transformadores afinados. Cuando se combinaron los distintos tipos de características, los resultados mejoraron aún más, mostrando que cada una aporta conocimientos complementarios sobre el comportamiento de los péptidos. Para sondear lo que el modelo había aprendido, los autores usaron SHAP, una técnica popular de IA explicable, para medir cómo diferentes características latentes influían en las predicciones. Aunque estas características son abstractas, el análisis confirmó que el modelo se apoya en un conjunto compacto de patrones discriminativos y biológicamente significativos en lugar de ruido aleatorio. En términos sencillos, el sistema parece “mirar” los tipos de señales correctas.
Qué implica esto para las medicinas futuras
Para no especialistas, la conclusión clave es que este trabajo proporciona un filtro basado en datos y altamente preciso para cribar enormes números de secuencias peptídicas y señalar aquellas con más probabilidades de actuar como agentes antimicrobianos o anticancerígenos eficaces. Al mezclar modelado generativo, múltiples modelos del lenguaje de proteínas y aprendizaje profundo explicable, GAC-BiTCNN-AMP ofrece una forma escalable de priorizar candidatos para pruebas de laboratorio, acelerando potencialmente el desarrollo de nuevos tratamientos contra infecciones y cánceres que resisten las terapias actuales. Extensiones futuras podrían no solo predecir qué péptidos funcionan, sino también guiar el diseño de secuencias completamente nuevas optimizadas para potencia, selectividad y seguridad.
Cita: Ali, F., Khalid, M., Alsini, R. et al. A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network. Sci Rep 16, 13801 (2026). https://doi.org/10.1038/s41598-026-43370-6
Palabras clave: péptidos antimicrobianos, modelos del lenguaje de proteínas, aprendizaje profundo, oncología de precisión, descubrimiento de fármacos