Clear Sky Science · nl
Een generatief verklaarbaar model voor voorspelling van antimicrobiële peptiden met bidirectionele temporele convolutionele neurale netwerken
Infecties en kanker bestrijden met slimme peptiden
Geneesmiddelen die gevaarlijke microben en kankercellen kunnen doden zonder gezond weefsel te schaden zijn een langgekoesterde ambitie in de geneeskunde. De natuur maakt zulke moleculen al: antimicrobiële peptiden, kleine eiwitfragmenten die gaten in membraan van microben slaan en tevens het immuunsysteem kunnen moduleren. Maar het vinden van de meest veelbelovende peptiden tussen het enorme aantal mogelijke aminozuurreeksen is als zoeken naar een speld in een hooiberg. Deze studie introduceert een krachtig kunstmatig-intelligentie-framework, GAC-BiTCNN-AMP, dat leert van grote biologische datasets om te voorspellen welke peptiden waarschijnlijk effectieve antimicrobiële middelen en potentiële kankertherapeutica zijn.
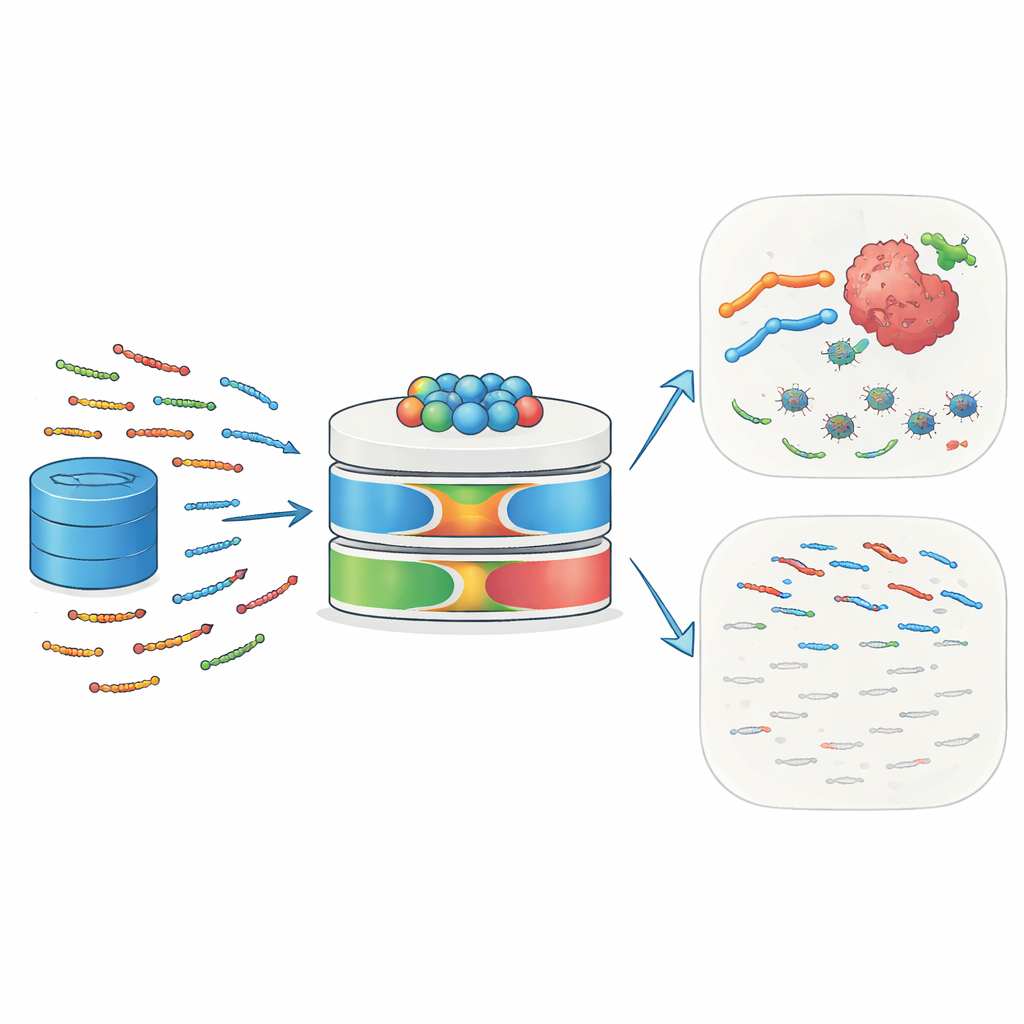
De kleine lijfwachten van de natuur
Antimicrobiële peptiden (AMPs) zijn korte ketens van 10 tot 50 aminozuren die voorkomen bij mensen, dieren, planten en microben. Ze fungeren als een eerste verdedigingslinie door zich te binden aan microbieel oppervlak, hun membranen te verstoren en celdood te veroorzaken. Veel AMPs rekruteren ook immuuncellen, beïnvloeden ontstekingsreacties en hervormen de lokale weefselomgeving. Kankercellen, met hun atypisch geladen en ongeordende membranen, kunnen bijzonder kwetsbaar zijn voor dergelijke peptiden. Sommige bekende voorbeelden — waaronder melittine en defensines — hebben aangetoond tumoren te kunnen doden, ze gevoeliger te maken voor chemotherapie of bestraling, en immuunresponsen tegen tumoren te stimuleren. Deze dubbele rol tegen infectie en kanker maakt AMPs aantrekkelijke kandidaten voor volgende generaties precisiegeneesmiddelen.
Waarom traditionele voorspellingsinstrumenten tekortschieten
Ondanks hun potentieel blijft het in silico identificeren van nieuwe AMPs lastig. Eerdere computermodellen vertrouwden voornamelijk op eenvoudige sequentiepatronen en klassieke machine-learningtechnieken. Ze negeerden vaak rijkere informatie over hoe aminozuren over lange afstanden in een eiwit met elkaar interageren, hoe deze sequenties evolueerden, en welke subtiele fysische kenmerken een peptide zowel actief als selectief maken. Veel modellen gebruikten beperkte of redundante trainingsdata, sloegen systematische feature-selectie over en boden weinig interpreteerbaarheid — onderzoekers konden niet eenvoudig zien welke aspecten van de input een voorspelling aandreven. Daardoor waren hun nauwkeurigheid en vermogen om te generaliseren naar nieuwe peptiden beperkt, en slaagden ze er niet in de diverse biologische rollen van AMPs goed te vangen.
Een rijker beeld opbouwen uit sequenties
Om deze leemtes te vullen, verzamelden de auteurs eerst een grote, zorgvuldig gefilterde dataset uit zes AMP-databases en UniProt. Ze onderscheiden actieve peptiden van inactieve op basis van strenge experimentele criteria en verminderden redundantie zodat nauwe verwantschap tussen sequenties de prestatiemeting niet zou vertekenen. Daarna transformeerden ze elke peptide-sequentie naar meerdere complementaire numerieke representaties. Drie state-of-the-art proteïne-taalmodellen — ProtTrans-T5, UniRep en ESM-2 — werden gebruikt om hoge-dimensionale embeddings te genereren die context, langafstandafhankelijkheden en evolutionaire patronen coderen die geleerd zijn uit miljoenen eiwitten. Een aangepast descriptor genaamd PsePSSM-DCT voegde informatie toe over hoe elke positie in een sequentie de neiging heeft te muteren in de evolutie en hoe die patronen soepel variëren langs de sequentie. Een feature-selectiestap gebaseerd op XGBoost distilleerde deze rijke representaties vervolgens tot de meest informatieve componenten, waarbij ruis werd teruggesnoeid en signaal behouden bleef.
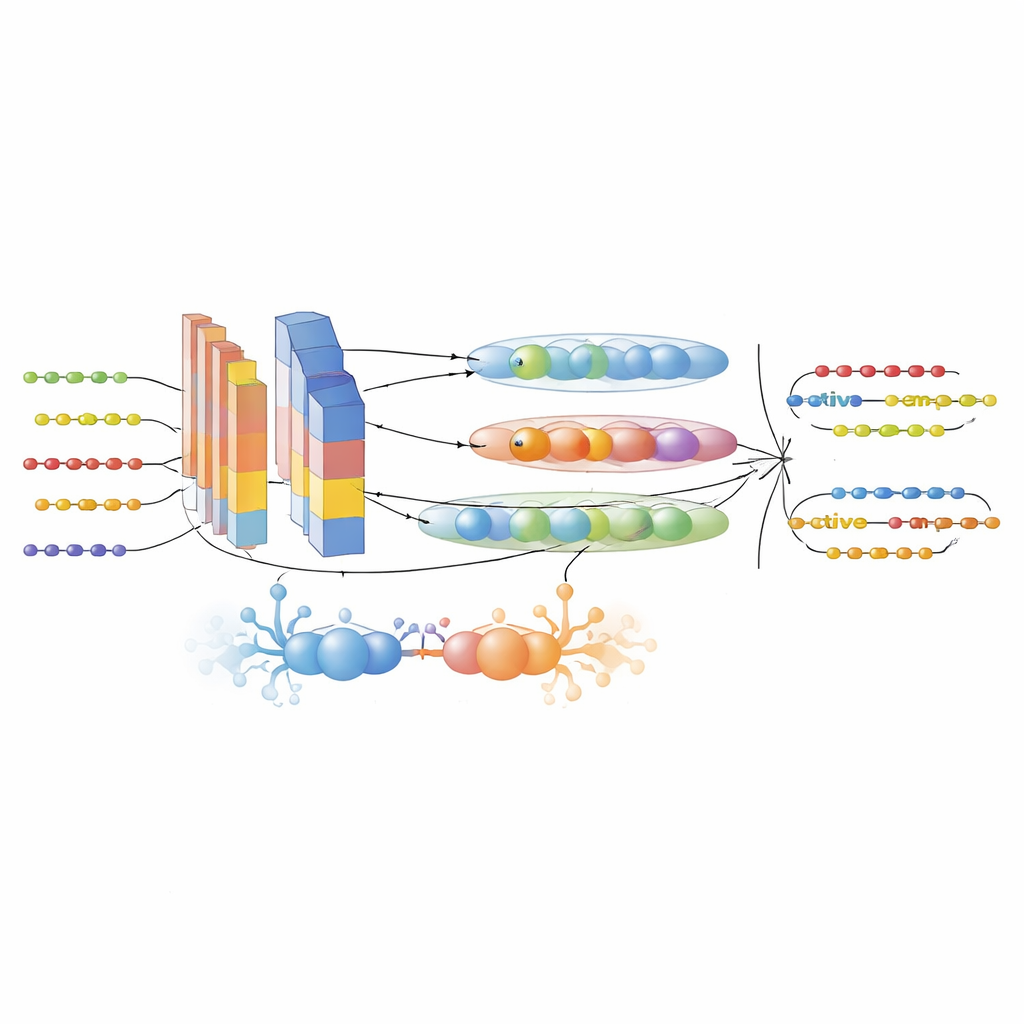
Een hybride AI-motor voor peptideontdekking
Het hart van het framework is het GAC-BiTCNN-model, een hybride deep-learningarchitectuur specifiek afgestemd op sequentiedata. Het combineert meerdere ideeën: een generatief adversarieel module die realistische synthetische featurevectoren creëert om de trainingsset te balanceren en te verrijken; convolutionele lagen die lokale motieven detecteren; een bidirectioneel temporeel convolutioneel netwerk dat patronen zowel vooruit als achteruit langs de sequentie vastlegt; en capsule-netwerken die gerelateerde kenmerken samenvoegen in kleine vectoriale "capsules" en hiërarchische relaties behouden. Elk type feature — taalmodel-embeddings en evolutionaire descriptors — wordt in een eigen stroom verwerkt en later samengevoegd. Het model werd getraind en afgestemd met cross-validatie en vervolgens getest op een volledig aparte, tijdgescheiden dataset van recentere peptide-invoer om information leakage te minimaliseren.
Prestaties, verklaarbaarheid en wat het betekent
GAC-BiTCNN-AMP behaalde opvallende prestaties: tot ongeveer 97% accuratesse en bijna perfecte AUC-scores in cross-validatie, en meer dan 95% accuratesse op de onafhankelijke testset, waarmee het een reeks bestaande AMP-voorspellers en zelfs alleen-fijn-afgestelde transformer-baselines overtrof. Wanneer de verschillende featuretypes werden gecombineerd, verbeterden de resultaten verder, wat aantoont dat elk complementaire kennis bijdraagt over peptidegedrag. Om te onderzoeken wat het model had geleerd, gebruikten de auteurs SHAP, een veelgebruikte verklaarbare-AI-techniek, om te meten hoe verschillende latente features voorspellingen beïnvloedden. Hoewel deze features abstract zijn, bevestigde de analyse dat het model vertrouwt op een compacte set discriminatieve, biologisch betekenisvolle patronen in plaats van willekeurige ruis. In gewone taal lijkt het systeem naar de juiste soorten signalen te "kijken".
Wat dit betekent voor toekomstige geneesmiddelen
Voor niet-specialisten is de belangrijkste conclusie dat dit werk een zeer nauwkeurig, data-gedreven filter biedt om door enorme aantallen peptide-sequenties te zoeken en die te identificeren die het meest waarschijnlijk als effectieve antimicrobiële of anti-kanker middelen zullen werken. Door generatief modelleren, meerdere proteïne-taalmodellen en verklaarbare deep learning te combineren, biedt GAC-BiTCNN-AMP een schaalbare manier om kandidaten te prioriteren voor laboratoriumtesten, wat mogelijk de ontwikkeling van nieuwe behandelingen tegen infecties en kankers die huidige therapieën weerstaan kan versnellen. Toekomstige uitbreidingen kunnen niet alleen voorspellen welke peptiden werken, maar ook het ontwerp van volledig nieuwe sequenties sturen die zijn afgestemd op potentie, selectiviteit en veiligheid.
Bronvermelding: Ali, F., Khalid, M., Alsini, R. et al. A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network. Sci Rep 16, 13801 (2026). https://doi.org/10.1038/s41598-026-43370-6
Trefwoorden: antimicrobiële peptiden, proteïne-taalmodellen, deep learning, precisie-oncologie, geneesmiddelenontwikkeling