Clear Sky Science · fr
Un modèle génératif explicable pour la prédiction des peptides antimicrobiens utilisant un réseau neuronal convolutif temporel bidirectionnel
Combattre les infections et le cancer avec des peptides intelligents
Des médicaments capables d’éliminer des microbes dangereux et des cellules cancéreuses sans endommager les tissus sains constituent un rêve de longue date en médecine. La nature produit déjà de telles molécules : les peptides antimicrobiens, de courts fragments protéiques qui perforent les membranes microbiennes et peuvent aussi moduler le système immunitaire. Mais repérer les peptides les plus prometteurs parmi l’immense nombre de séquences d’acides aminés possibles revient à chercher une aiguille dans une meule de foin. Cette étude présente un puissant cadre d’intelligence artificielle, GAC-BiTCNN-AMP, qui apprend à partir de larges jeux de données biologiques pour prédire quels peptides sont susceptibles d’être des agents antimicrobiens efficaces et de potentiels thérapeutiques anticancéreux.
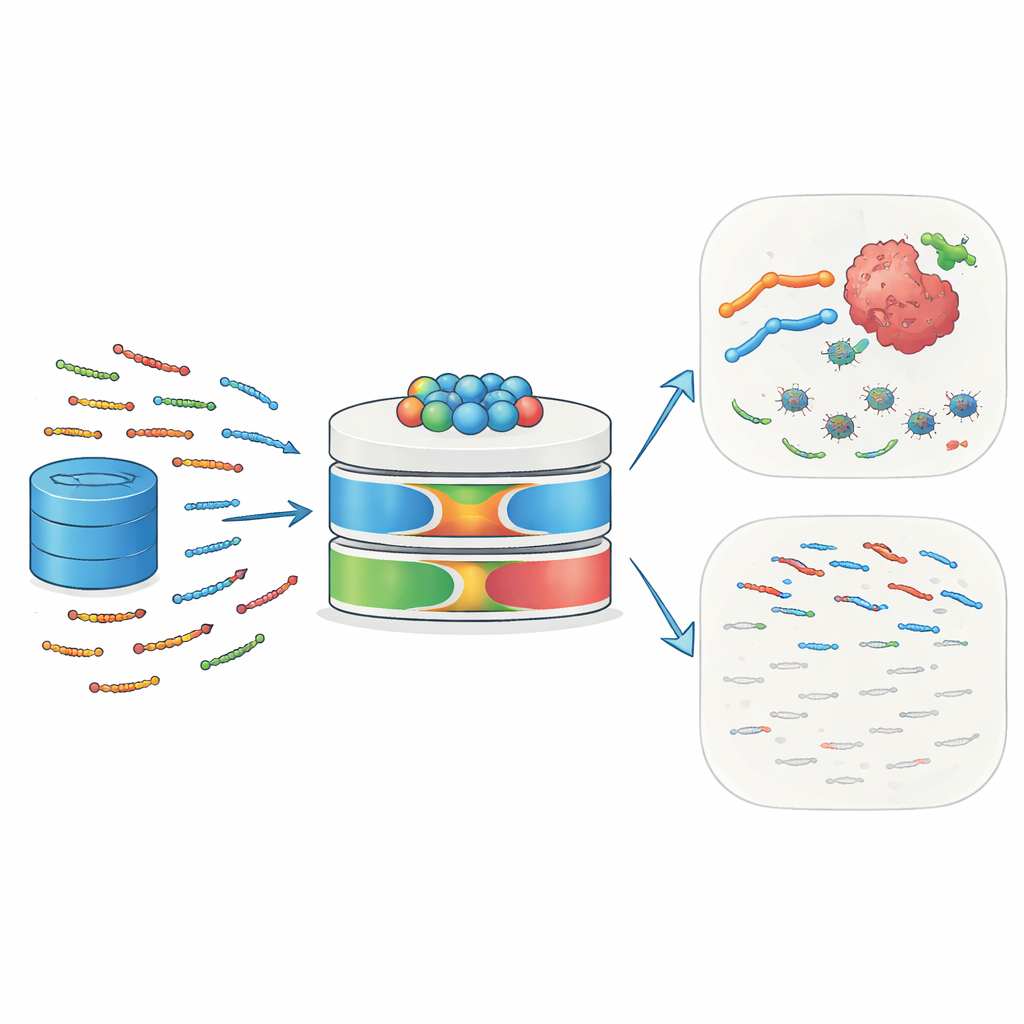
Les petits gardiens de la nature
Les peptides antimicrobiens (PAM) sont de courtes chaînes de 10 à 50 acides aminés présentes chez l’homme, les animaux, les plantes et les microbes. Ils constituent une première ligne de défense en se fixant aux surfaces microbiennes, en perturbant leurs membranes et en déclenchant la mort cellulaire. De nombreux PAM recrutent également des cellules immunitaires, influencent l’inflammation et modifient l’environnement tissulaire local. Les cellules cancéreuses, avec leurs membranes souvent chargées différemment et désordonnées, peuvent être particulièrement vulnérables à ces peptides. Quelques exemples bien connus — notamment la mélittine et les défensines — ont montré qu’ils pouvaient tuer des cellules tumorales, les sensibiliser à la chimiothérapie ou à la radiothérapie, et stimuler des réponses immunitaires contre les tumeurs. Ce double rôle contre l’infection et le cancer fait des PAM des candidats attractifs pour des médicaments de précision de nouvelle génération.
Pourquoi les outils traditionnels de prédiction sont insuffisants
Malgré leur potentiel, l’identification de nouveaux PAM in silico reste difficile. Les modèles informatiques antérieurs s’appuyaient principalement sur des motifs simples de séquence et des techniques d’apprentissage automatique classiques. Ils ignoraient souvent des informations plus riches sur la manière dont les acides aminés interagissent à longue distance dans une protéine, sur l’évolution de ces séquences, et sur quelles caractéristiques physiques subtiles rendent un peptide à la fois actif et sélectif. De nombreux modèles utilisaient des jeux de données limités ou redondants, omettaient une sélection systématique des caractéristiques et offraient peu d’interprétabilité — les chercheurs ne pouvaient pas facilement voir quels aspects de l’entrée guidaient une prédiction. En conséquence, leur précision et leur capacité à généraliser à de nouveaux peptides étaient limitées, et ils peinaient à capturer la diversité des rôles biologiques que peuvent jouer les PAM.
Construire une image plus riche à partir des séquences
Pour combler ces lacunes, les auteurs ont d’abord rassemblé un grand jeu de données soigneusement filtré à partir de six bases de données de PAM et d’UniProt. Ils ont distingué les peptides actifs des inactifs selon des critères expérimentaux stricts et réduit la redondance afin que des séquences étroitement apparentées n’influent pas artificiellement sur les performances. Ensuite, ils ont transformé chaque séquence peptidique en plusieurs vues numériques complémentaires. Trois modèles de langage pour protéines à la pointe — ProtTrans-T5, UniRep et ESM-2 — ont été utilisés pour générer des embeddings de haute dimension qui codent le contexte, les dépendances à longue portée et les schémas évolutifs appris à partir de millions de protéines. Un descripteur personnalisé nommé PsePSSM-DCT a ajouté des informations sur la propension de chaque position de la séquence à muter au cours de l’évolution et sur la manière dont ces motifs varient de façon lisse le long de la séquence. Une étape de sélection de caractéristiques basée sur XGBoost a ensuite distillé ces représentations riches vers les composantes les plus informatives, réduisant le bruit tout en préservant le signal.
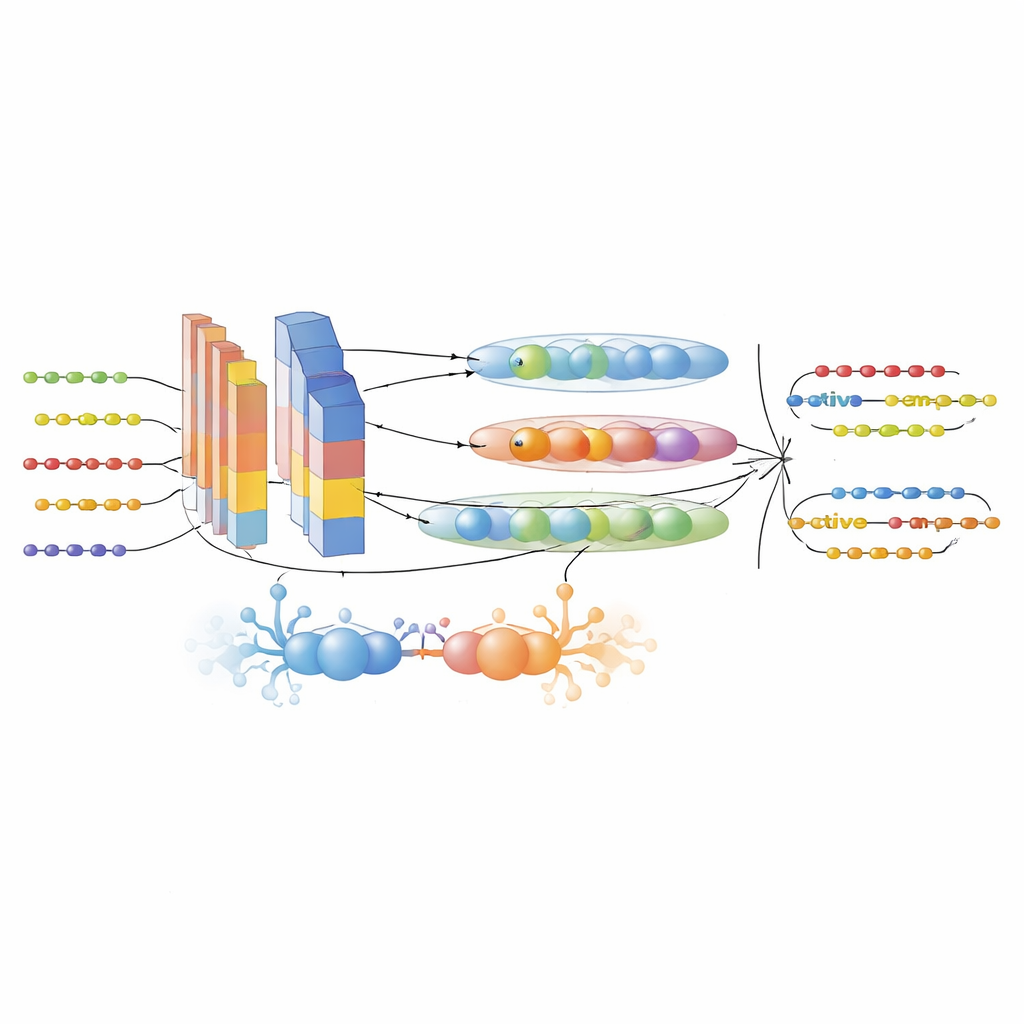
Un moteur d’IA hybride pour la découverte de peptides
Le cœur du cadre est le modèle GAC-BiTCNN, une architecture d’apprentissage profond hybride spécifiquement adaptée aux données de séquence. Il combine plusieurs idées : un module génératif adversarial qui crée des vecteurs de caractéristiques synthétiques réalistes pour équilibrer et enrichir le jeu d’entraînement ; des couches convolutionnelles qui détectent des motifs locaux ; un réseau convolutionnel temporel bidirectionnel qui capture des motifs s’étendant dans les deux sens le long de la séquence ; et des réseaux à capsules qui regroupent des caractéristiques liées en petites « capsules » vectorielles, préservant les relations hiérarchiques. Chaque type de caractéristique — embeddings issus des modèles de langage et descripteurs évolutionnaires — est traité dans son propre flux puis fusionné par la suite. Le modèle a été entraîné et optimisé par validation croisée puis testé sur un jeu de données entièrement séparé et séparé dans le temps, composé d’entrées de peptides plus récentes, afin de minimiser les fuites d’information.
Performances, interprétabilité et signification
GAC-BiTCNN-AMP a atteint des performances remarquables : jusqu’à environ 97 % de précision et des scores aire sous la courbe quasi parfaits en validation croisée, et plus de 95 % de précision sur le jeu de test indépendant, dépassant une série de prédicteurs PAM existants et même des baselines composés uniquement de transformeurs ajustés. Lorsque les différents types de caractéristiques étaient combinés, les résultats s’amélioraient encore, montrant que chacun apporte une connaissance complémentaire sur le comportement des peptides. Pour sonder ce que le modèle avait appris, les auteurs ont utilisé SHAP, une technique d’IA explicable populaire, pour mesurer comment différentes caractéristiques latentes influaient sur les prédictions. Bien que ces caractéristiques soient abstraites, l’analyse a confirmé que le modèle s’appuie sur un ensemble compact de motifs discriminants et biologiquement significatifs plutôt que sur du bruit aléatoire. En termes simples, le système semble « regarder » les bons types de signaux.
Ce que cela signifie pour les médicaments de demain
Pour les non-spécialistes, la conclusion clé est que ce travail fournit un filtre fondé sur les données, hautement précis, pour trier d’énormes nombres de séquences peptidiques et identifier celles les plus susceptibles d’agir comme agents antimicrobiens ou anticancéreux efficaces. En combinant modélisation générative, plusieurs modèles de langage pour protéines et apprentissage profond explicable, GAC-BiTCNN-AMP offre une manière évolutive de prioriser des candidats pour des tests en laboratoire, accélérant potentiellement le développement de nouveaux traitements contre les infections et les cancers résistants aux thérapies actuelles. Des extensions futures pourraient non seulement prédire quels peptides fonctionnent, mais aussi guider la conception de séquences entièrement nouvelles optimisées pour la puissance, la sélectivité et la sécurité.
Citation: Ali, F., Khalid, M., Alsini, R. et al. A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network. Sci Rep 16, 13801 (2026). https://doi.org/10.1038/s41598-026-43370-6
Mots-clés: peptides antimicrobiens, modèles de langage pour protéines, apprentissage profond, oncologie de précision, découverte de médicaments