Clear Sky Science · de
Ein generatives erklärbares Modell zur Vorhersage antimikrobieller Peptide mithilfe bidirektionaler temporaler Faltungsnetzwerke
Infektionen und Krebs mit intelligenten Peptiden bekämpfen
Medikamente, die gefährliche Mikroben und Krebszellen abtöten, ohne gesundes Gewebe zu schädigen, sind ein lange gehegter Traum der Medizin. Die Natur stellt solche Moleküle bereits her: antimikrobielle Peptide, winzige Proteinfragmente, die Löcher in mikrobielle Membranen schlagen und zudem das Immunsystem modulieren können. Unter der Vielzahl möglicher Aminosäuresequenzen die vielversprechendsten Peptide zu finden, gleicht jedoch der Suche nach Nadeln im Heuhaufen. Diese Studie stellt ein leistungsfähiges KI-Framework vor, GAC-BiTCNN-AMP, das aus großen biologischen Datensätzen lernt, um vorherzusagen, welche Peptide wahrscheinlich wirksame antimikrobielle Wirkstoffe und potenzielle Krebsmedikamente sind.
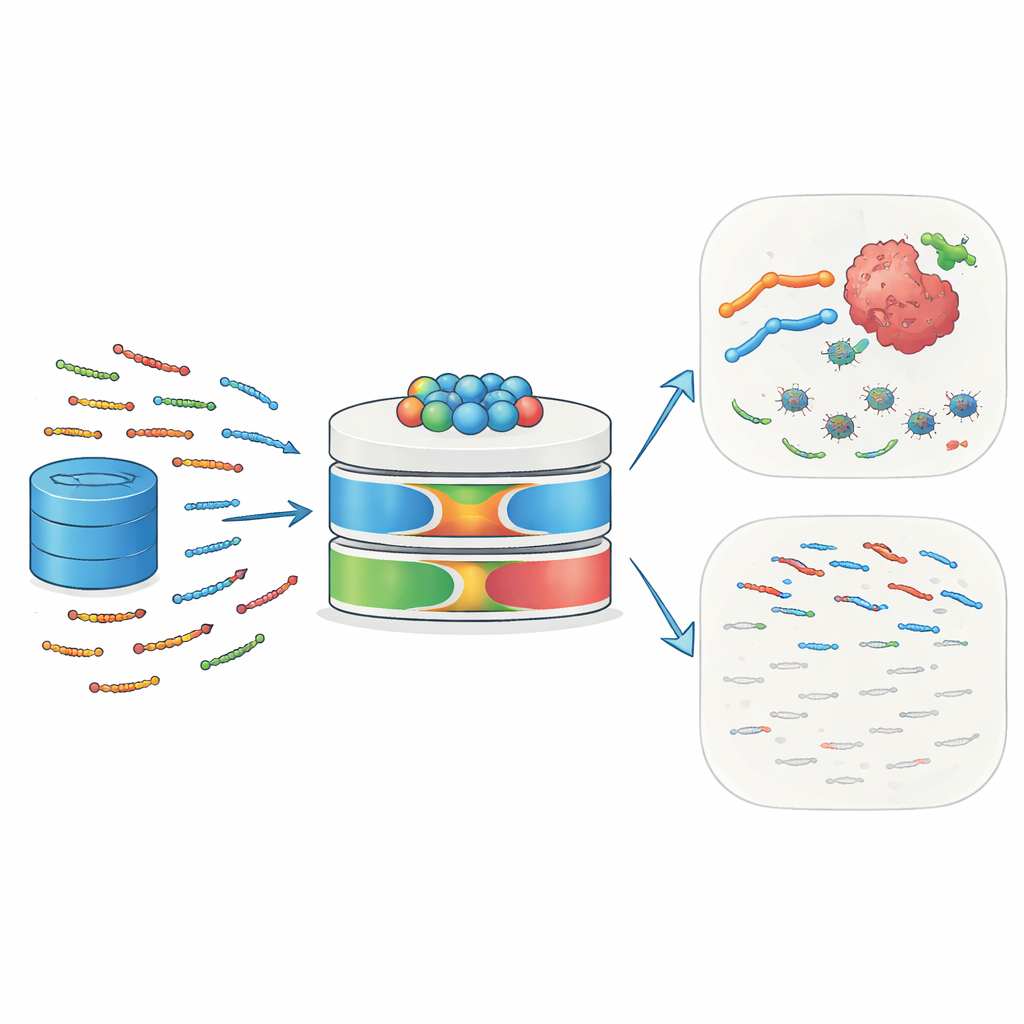
Natur’s winzige Bodyguards
Antimikrobielle Peptide (AMPs) sind kurze Ketten von 10 bis 50 Aminosäuren, die bei Menschen, Tieren, Pflanzen und Mikroben vorkommen. Sie wirken als erste Verteidigungslinie, indem sie an mikrobielle Oberflächen binden, deren Membranen stören und den Zelltod auslösen. Viele AMPs rekrutieren zudem Immunzellen, beeinflussen Entzündungsprozesse und verändern die lokale Gewebeumgebung. Krebszellen sind aufgrund ihrer ungewöhnlichen Ladungsverhältnisse und der unstrukturierten Membranzusammensetzung besonders empfindlich gegenüber solchen Peptiden. Einige bekannte Beispiele – darunter Melittin und Defensine – konnten Tumorzellen abtöten, diese für Chemotherapie oder Strahlung sensibilisieren und Immunantworten gegen Tumoren stimulieren. Diese doppelte Wirkung gegen Infektionen und Krebs macht AMPs zu attraktiven Kandidaten für die nächste Generation präziser Medikamente.
Warum traditionelle Vorhersagewerkzeuge nicht ausreichen
Trotz ihres Potenzials bleibt die Identifizierung neuer AMPs in silico schwierig. Frühere Computermodelle stützten sich vorwiegend auf einfache Sequenzmuster und klassische Machine-Learning-Verfahren. Häufig ignorierten sie reichere Informationen darüber, wie Aminosäuren über lange Distanzen in einem Protein interagieren, wie diese Sequenzen evolvierten und welche subtilen physikalischen Merkmale ein Peptid sowohl wirksam als auch selektiv machen. Viele Modelle nutzten eingeschränkte oder redundante Trainingsdaten, übersprangen systematische Merkmalsauswahl und boten geringe Interpretierbarkeit – Forschende konnten nicht leicht erkennen, welche Aspekte der Eingabe eine Vorhersage beeinflussten. In der Folge waren Genauigkeit und Generalisierungsfähigkeit auf neue Peptide begrenzt, und es fiel ihnen schwer, die vielfältigen biologischen Rollen von AMPs abzubilden.
Ein reichhaltigeres Bild aus Sequenzen aufbauen
Um diese Lücken zu schließen, stellten die Autorinnen und Autoren zunächst einen großen, sorgfältig gefilterten Datensatz aus sechs AMP-Datenbanken und UniProt zusammen. Sie unterschieden aktive Peptide von inaktiven anhand strenger experimenteller Kriterien und reduzierten Redundanz, damit eng verwandte Sequenzen die Leistung nicht künstlich aufblähen. Anschließend wandeln sie jede Peptidsequenz in mehrere komplementäre numerische Darstellungen um. Drei moderne Protein-Sprachmodelle – ProtTrans-T5, UniRep und ESM-2 – erzeugten hochdimensionale Einbettungen, die Kontext, Fernabhängigkeiten und evolutionäre Muster kodieren, die aus Millionen von Proteinen gelernt wurden. Ein kundenspezifischer Deskriptor namens PsePSSM-DCT fügte Informationen darüber hinzu, wie sich Positionen einer Sequenz in der Evolution zu verändern tendieren und wie diese Muster sich entlang der Sequenz glatt variieren. Ein Merkmalsauswahl-Schritt auf Basis von XGBoost destillierte diese reichen Repräsentationen zu den informativsten Komponenten, reduzierte Rauschen und bewahrte gleichzeitig das Signal.
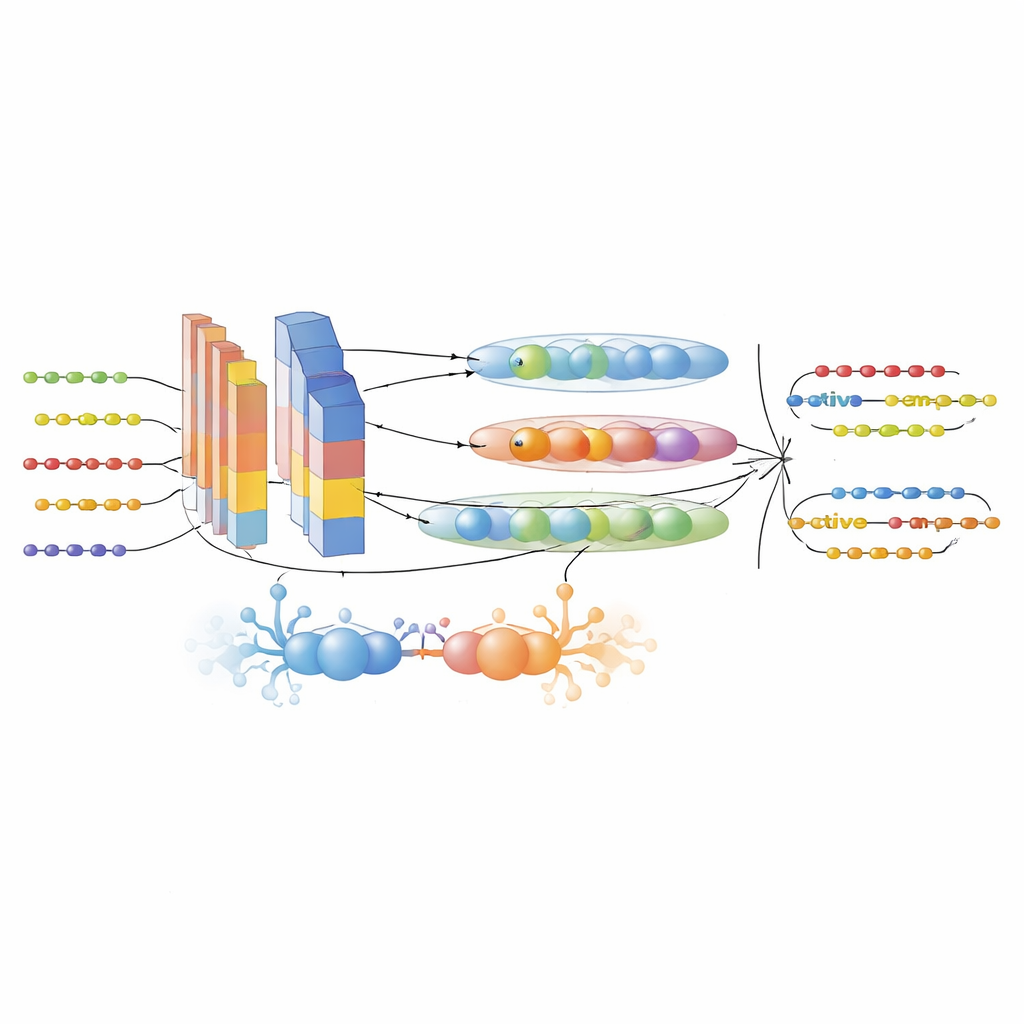
Eine hybride KI‑Engine zur Peptidentdeckung
Das Herzstück des Frameworks ist das GAC-BiTCNN-Modell, eine hybride Deep-Learning-Architektur, die speziell für Sequenzdaten zugeschnitten ist. Sie kombiniert mehrere Konzepte: ein generatives gegnerisches Modul, das realistische synthetische Merkmalsvektoren erzeugt, um den Trainingssatz auszugleichen und anzureichern; Faltungsschichten, die lokale Motive erkennen; ein bidirektionales temporales Faltungsnetzwerk, das Muster sowohl vorwärts als auch rückwärts entlang der Sequenz einfängt; und Kapselnetzwerke, die verwandte Merkmale in kleine Vektor-„Kapseln“ gruppieren und hierarchische Beziehungen bewahren. Jeder Merkmalstyp – Sprachmodell-Einbettungen und evolutionäre Deskriptoren – wird in einem eigenen Strom verarbeitet und später fusioniert. Das Modell wurde mittels Kreuzvalidierung trainiert und abgestimmt und anschließend an einem vollständig separaten, zeitlich getrennten Testsatz neuerer Peptid-Einträge geprüft, um Informationsleckage zu minimieren.
Leistung, Erklärbarkeit und Bedeutung
GAC-BiTCNN-AMP erzielte herausragende Leistungen: bis zu rund 97 % Genauigkeit und nahezu perfekte AUC-Werte in der Kreuzvalidierung sowie über 95 % Genauigkeit im unabhängigen Testsatz und übertraf damit eine Reihe bestehender AMP-Prädiktoren und sogar feinabgestimmte Transformer‑Alleinmodelle. Wenn die verschiedenen Merkmalstypen kombiniert wurden, verbesserten sich die Ergebnisse weiter, was zeigt, dass jeder Typ komplementäre Informationen über das Verhalten von Peptiden beiträgt. Um zu untersuchen, was das Modell gelernt hat, nutzten die Autorinnen und Autoren SHAP, eine verbreitete Explainable-AI‑Technik, um zu messen, wie unterschiedliche latente Merkmale Vorhersagen beeinflussen. Zwar sind diese Merkmale abstrakt, die Analyse bestätigte jedoch, dass das Modell sich auf eine kompakte Menge diskriminativer, biologisch sinnträchtiger Muster stützt und nicht auf zufälliges Rauschen. Vereinfacht gesagt scheint das System auf die richtigen Signale zu schauen.
Was das für zukünftige Medikamente bedeutet
Für Nichtfachleute ist die wichtigste Erkenntnis, dass diese Arbeit einen hochgenauen, datengetriebenen Filter bereitstellt, um aus einer riesigen Anzahl von Peptidsequenzen diejenigen herauszufiltern, die am wahrscheinlichsten als wirksame antimikrobielle oder antitumorale Wirkstoffe fungieren. Durch die Kombination von generativem Modeling, mehreren Protein-Sprachmodellen und erklärbarem Deep Learning bietet GAC-BiTCNN-AMP einen skalierbaren Weg, Kandidaten für Labortests zu priorisieren und so die Entwicklung neuer Therapien gegen Infektionen und Krebserkrankungen, die gegen aktuelle Behandlungen resistent sind, zu beschleunigen. Zukünftige Erweiterungen könnten nicht nur vorhersagen, welche Peptide wirken, sondern auch die Gestaltung völlig neuer Sequenzen lenken, die auf Wirksamkeit, Selektivität und Sicherheit abgestimmt sind.
Zitation: Ali, F., Khalid, M., Alsini, R. et al. A generative explainable model for antimicrobial peptide prediction using bidirectional temporal convolutional neural network. Sci Rep 16, 13801 (2026). https://doi.org/10.1038/s41598-026-43370-6
Schlüsselwörter: antimikrobielle Peptide, Protein-Sprachmodelle, Deep Learning, Präzisionsonkologie, Arzneimittelentdeckung