Clear Sky Science · pl
Wpływ mutacji PD-1 na wiązanie pembrolizumabu: wnioski z symulacji dynamiki molekularnej i analizy MM-GBSA
Dlaczego to ma znaczenie dla leczenia raka
Leki immunoterapeutyczne, takie jak pembrolizumab, zrewolucjonizowały opiekę nad wieloma nowotworami, jednak tylko część pacjentów odnosi korzyść — a niektórzy, którzy początkowo reagują, później relapsują. W tym badaniu zadano konkretne pytanie kryjące się za tą kliniczną zagadką: co się dzieje, gdy w PD-1 — molekularnym „wyłączniku” układu odpornościowego, który ma blokować pembrolizumab — pojawią się subtelne zmiany? Korzystając z zaawansowanych symulacji komputerowych zamiast próbek pacjentów, autorzy mapują, jak rzadkie zmiany genetyczne w PD-1 mogą osłabić lub wzmocnić przyczepność leku, dostarczając wskazówek, dlaczego leczenie czasem zawodzi i jak przyszłe testy lub leki mogłyby działać lepiej.
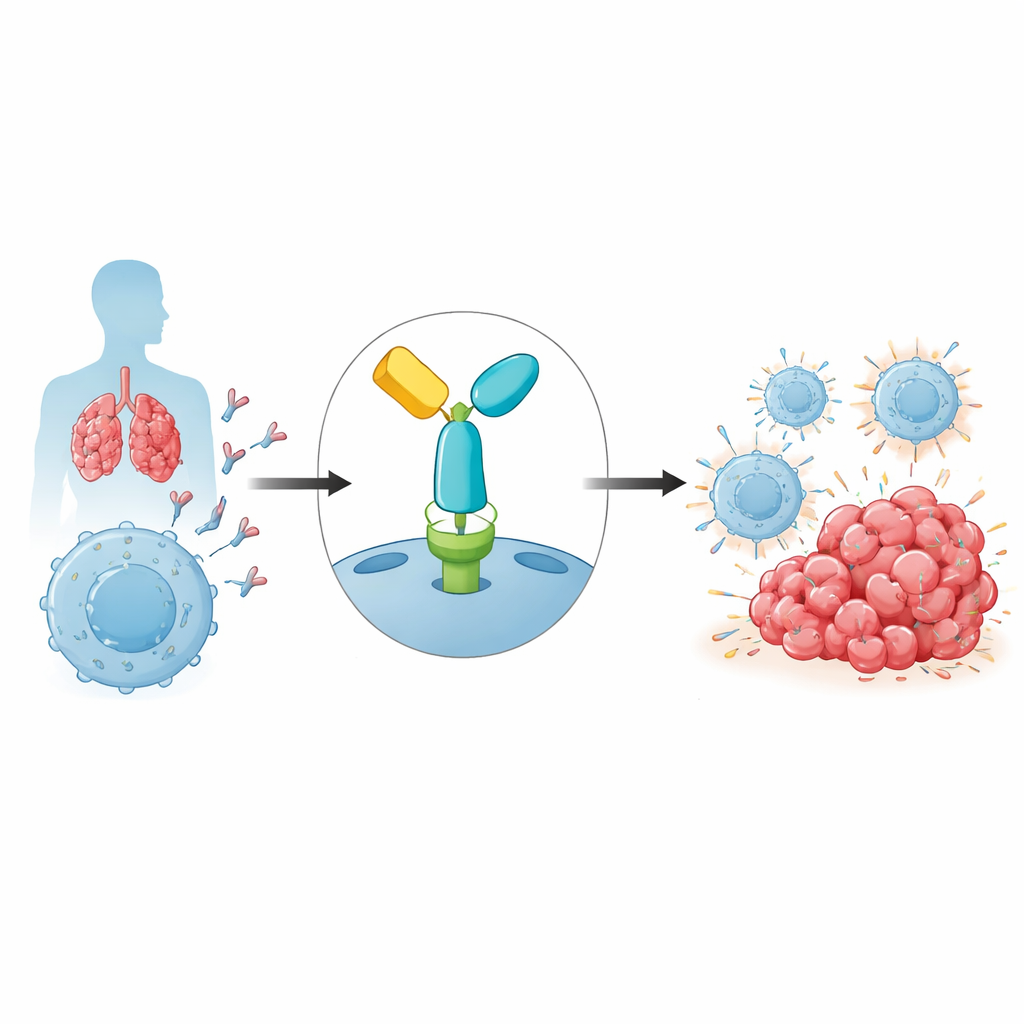
Hamulce odporności, których wykorzystują nowotwory
Nasz układ odpornościowy polega na limfocytach T, które rozpoznają i atakują nieprawidłowe komórki, w tym nowotworowe. Aby uniknąć szkód ubocznych, limfocyty T mają bezpieczniki zwane punktami kontrolnymi. Jednym z najważniejszych jest PD-1, receptor na limfocytach T, który po związaniu z ligandem PD-L1 na innych komórkach tłumi aktywność odpornościową. Nowotwory często wykorzystują tę ścieżkę, eksponując PD-L1, skutecznie nakazując pobliskim limfocytom T wycofanie się. Pembrolizumab to przeciwciało, które wiąże PD-1 i blokuje tę hamującą interakcję, reaktywując limfocyty T, by mogły skuteczniej atakować komórki nowotworowe. Choć ta strategia doprowadziła do długotrwałych remisji w nowotworach takich jak czerniak czy rak płuca, oporność jest powszechna, a jej przyczyny wciąż są odsłaniane.
Komputery jako molekularny mikroskop
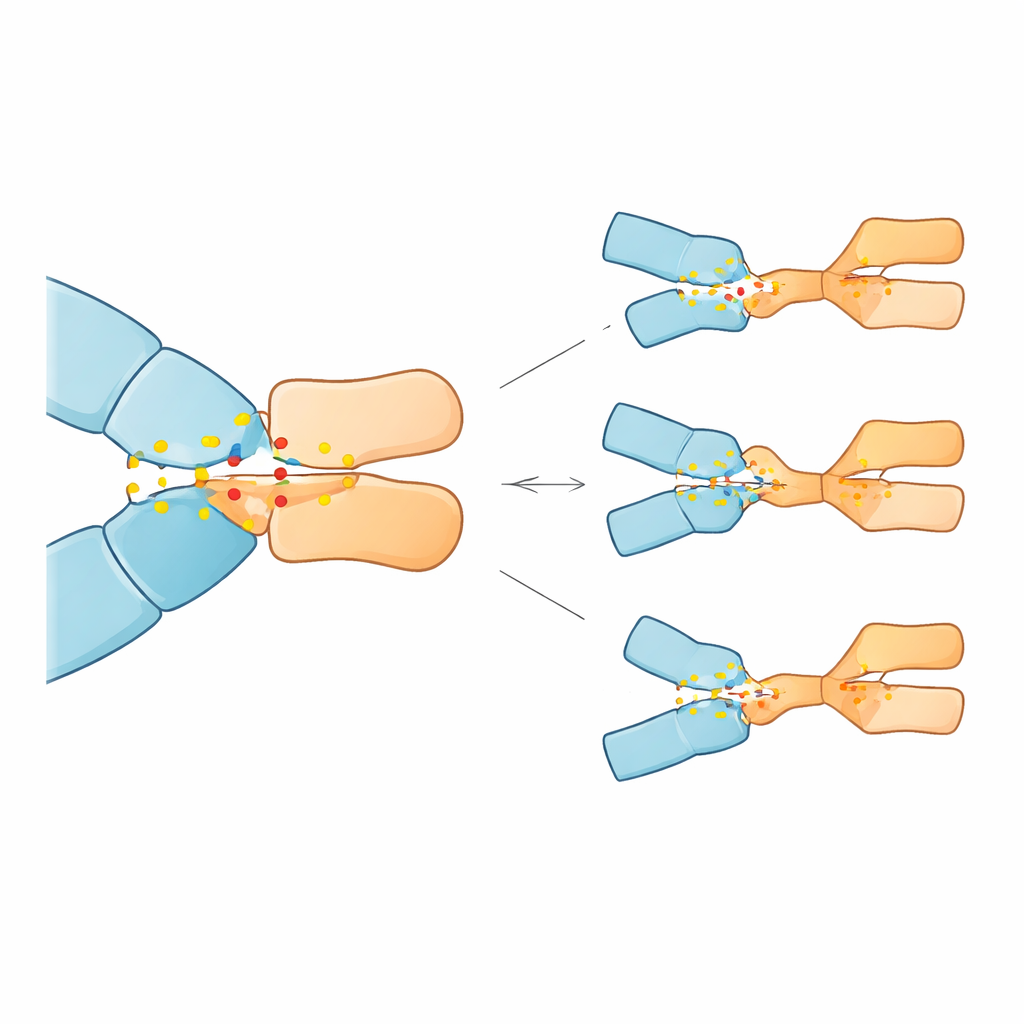
Zamiast przeprowadzać eksperymenty w laboratorium na komórkach, badacze sięgnęli po szczegółowe modele komputerowe białka PD-1 związanego z pembrolizumabem. Rozpoczęli od wysokorozdzielczej struktury krystalicznej tego kompleksu i zidentyfikowali kluczowe punkty styku — pojedyncze aminokwasy w PD-1, które tworzą wiązania wodorowe, hydrofobowe kontakty przypominające „smar”, lub jonowe „mostki solne” z lekiem. Następnie, używając symulacji dynamiki molekularnej, śledzili zachowanie tych kontaktów w skali 50 miliardowych części sekundy i obliczyli, ile każdy resztkowy aminokwas wnosi do utrzymania integralności kompleksu. Pozwoliło im to wskazać 16 „gorących punktów” w PD-1, które są szczególnie ważne dla silnego wiązania.
Rzadkie zmiany genetyczne o dużym znaczeniu
Następnie zespół przeszukał dużą bazę danych białek w poszukiwaniu naturalnie występujących zmian (mutacji) w tych gorących punktach i znalazł 40 wariantów oraz jedną dodatkową mutację wcześniej opisaną w badaniach laboratoryjnych jako zakłócająca wiązanie pembrolizumabu. Po odfiltrowaniu wariantów przewidywanych jako niszczące ogólną strukturę PD-1 pozostało 28 mutacji „prawdopodobnie łagodnych” — zmian, które powinny pozostawić PD-1 funkcjonalnym, ale mogą subtelnie zmienić jego interakcję z lekami. Dla każdej z tych mutacji zbudowali trójwymiarowy model mutanta i przeprowadzili tego samego rodzaju symulację dynamiki molekularnej, a następnie obliczenie energii metodą MM-GBSA, aby oszacować, jak silnie pembrolizumab wiązałby się w porównaniu z wersją normalną.
Gdy tracone są kluczowe kotwice
Symulacje wykazały, że 23 z 28 mutacji osłabiły wiązanie pembrolizumabu, niektóre bardzo znacząco, podczas gdy pięć je wzmocniło. Trzy mutacje wyróżniały się największym przewidywanym spadkiem powinowactwa: podstawienia w pozycjach 85 i 89 PD-1 (Asp85Gly, Asp85Asn i Pro89Arg). W białku normalnym Asp85 działa jako kotwica elektrostatyczna, tworząc trwały mostek solny i wiązania wodorowe z argininą na przeciwciele; Pro89 pomaga tworzyć klaster ciasnych, hydrofobowych kontaktów z kilkoma resztami przeciwciała. Mutacje Asp85 usunęły ładunek ujemny lub zdolność do tworzenia wiązań wodorowych w tym miejscu, skutecznie łamiąc kotwicę i destabilizując otaczającą sieć kontaktów. Zmiana Pro89Arg zastąpiła obojętną, dopasowującą kształt resztę aminokwasową resztą dodatkowo naładowaną dodatnio, co zaburzyło hydrofobowy klaster i zmniejszyło jego stabilizujący wkład. Dla kontrastu, kilka innych mutacji w różnych pozycjach subtelnie przekształciło interfejs w sposób, który w modelu mógł zwiększać wiązanie.
Co to może znaczyć dla pacjentów
Te wyniki sugerują, że nawet bardzo rzadkie zmiany w genie PD-1, jeśli pojawią się w kluczowych punktach styku, mogą zmniejszyć zdolność pembrolizumabu do przyczepienia się do celu i tym samym osłabić jego zdolność do uwolnienia limfocytów T przeciwko nowotworowi. Ponieważ praca ma charakter wyłącznie obliczeniowy i wymaga potwierdzenia eksperymentalnego oraz klinicznego, oferuje jednak mapę drogową: konkretne warianty PD-1, takie jak te w pozycjach 85 i 89, mogłyby kiedyś służyć jako biomarkery wskazujące pacjentów z wyższym ryzykiem oporności lub pomagać w doborze między różnymi lekami celującymi w PD-1. Szerzej rzecz biorąc, badanie ilustruje, jak pogłębione symulacje komputerowe mogą wyjaśniać molekularne przyczyny, dla których silne immunoterapie u niektórych osób odnoszą sukces, a u innych zawodzą.
Cytowanie: Bouricha, E.M., Hakmi, M., Batlamous, B. et al. The impact of PD-1 mutations on pembrolizumab binding: insights from molecular dynamics and MM-GBSA analysis. Sci Rep 16, 10773 (2026). https://doi.org/10.1038/s41598-026-45077-0
Słowa kluczowe: mutacje PD-1, oporność na pembrolizumab, immunoterapia przeciwnowotworowa, inhibitory punktów kontrolnych układu odpornościowego, symulacja dynamiki molekularnej