Clear Sky Science · nl
De impact van PD-1-mutaties op pembrolizumab-binding: inzichten uit moleculaire dynamica en MM-GBSA-analyse
Waarom dit belangrijk is voor de behandeling van kanker
Immunotherapiegeneesmiddelen zoals pembrolizumab hebben de zorg voor veel kankersoorten veranderd, maar slechts een deel van de patiënten profiteert ervan — en sommige patiënten die aanvankelijk reageren, krijgen later een terugval. Deze studie richt zich op een concreet vraagstuk achter dat klinische raadsel: wat gebeurt er als er subtiele veranderingen optreden in PD-1, de immuun "uit-schakelaar" die pembrolizumab probeert te blokkeren? Door geavanceerde computersimulaties te gebruiken in plaats van patiëntmateriaal, brengen de auteurs in kaart hoe zeldzame genetische veranderingen in PD-1 de hechting van het geneesmiddel kunnen verzwakken of versterken, en leveren ze aanwijzingen waarom de behandeling soms faalt en hoe toekomstige tests of geneesmiddelen het beter zouden kunnen doen.
De immuunrem die tumoren benutten
Ons immuunsysteem vertrouwt op T-cellen om afwijkende cellen, waaronder kankercellen, te herkennen en aan te vallen. Om collaterale schade te voorkomen, hebben T-cellen veiligheidschakelaars, zogenaamde immuuncheckpoints. Een van de belangrijkste is PD-1, een receptor op T-cellen die, wanneer deze wordt geactiveerd door zijn partner PD-L1 op andere cellen, de immuunactiviteit dempt. Tumoren maken vaak gebruik van dit pad door PD-L1 te tonen en als het ware tegen nabije T-cellen te zeggen dat ze zich terug moeten trekken. Pembrolizumab is een monoklonaal antilichaam dat aan PD-1 bindt en deze remmende interactie blokkeert, waardoor T-cellen weer geactiveerd worden zodat ze tumorcellen effectiever kunnen aanvallen. Hoewel deze strategie heeft geleid tot langdurige remissies bij kanker zoals melanoom en longkanker, komt resistentie veel voor en worden de onderliggende redenen nog steeds ontrafeld.
Computers als moleculair vergrootglas
In plaats van laboratoriumexperimenten op cellen uit te voeren, gebruikten de onderzoekers gedetailleerde computermodellen van het PD-1-eiwit in complex met pembrolizumab. Ze startten met een hogeresolutie-kristalstructuur van dit complex en identificeerden de belangrijke contactpunten — individuele aminozuren op PD-1 die waterstofbruggen, hydrofobe contacten of ionische "zoutbruggen" met het geneesmiddel vormen. Vervolgens volgden ze met moleculaire dynamica-simulaties hoe deze contacten zich over 50 nanoseconden (50 miljardsten van een seconde) gedroegen en berekenden ze hoeveel elk residu bijdroeg aan het intact houden van het complex. Dit stelde hen in staat 16 "hotspot"-posities op PD-1 te identificeren die vooral belangrijk zijn voor sterke binding.
Zeldzame genetische veranderingen met grote effecten
Vervolgens doorzocht het team een grote eiwitdatabase naar van nature voorkomende veranderingen (mutaties) in die hotspot-posities en vond 40 varianten, plus één extra mutatie die eerder in laboratoriumtests werd gerapporteerd als verstorend voor pembrolizumab-binding. Na het filteren van varianten die naar verwachting de algehele structuur van PD-1 zouden beschadigen, hielden ze 28 "waarschijnlijk goedaardige" mutaties over — veranderingen die PD-1 functioneel zouden moeten laten, maar subtiel kunnen beïnvloeden hoe het met geneesmiddelen interageert. Voor elk van deze bouwden ze een driedimensionaal gemuteerd model en voerden ze hetzelfde type moleculaire dynamica-simulatie uit, gevolgd door een energieberekeningsmethode (MM-GBSA) om te schatten hoe sterk pembrolizumab zou binden in vergelijking met de normale versie.
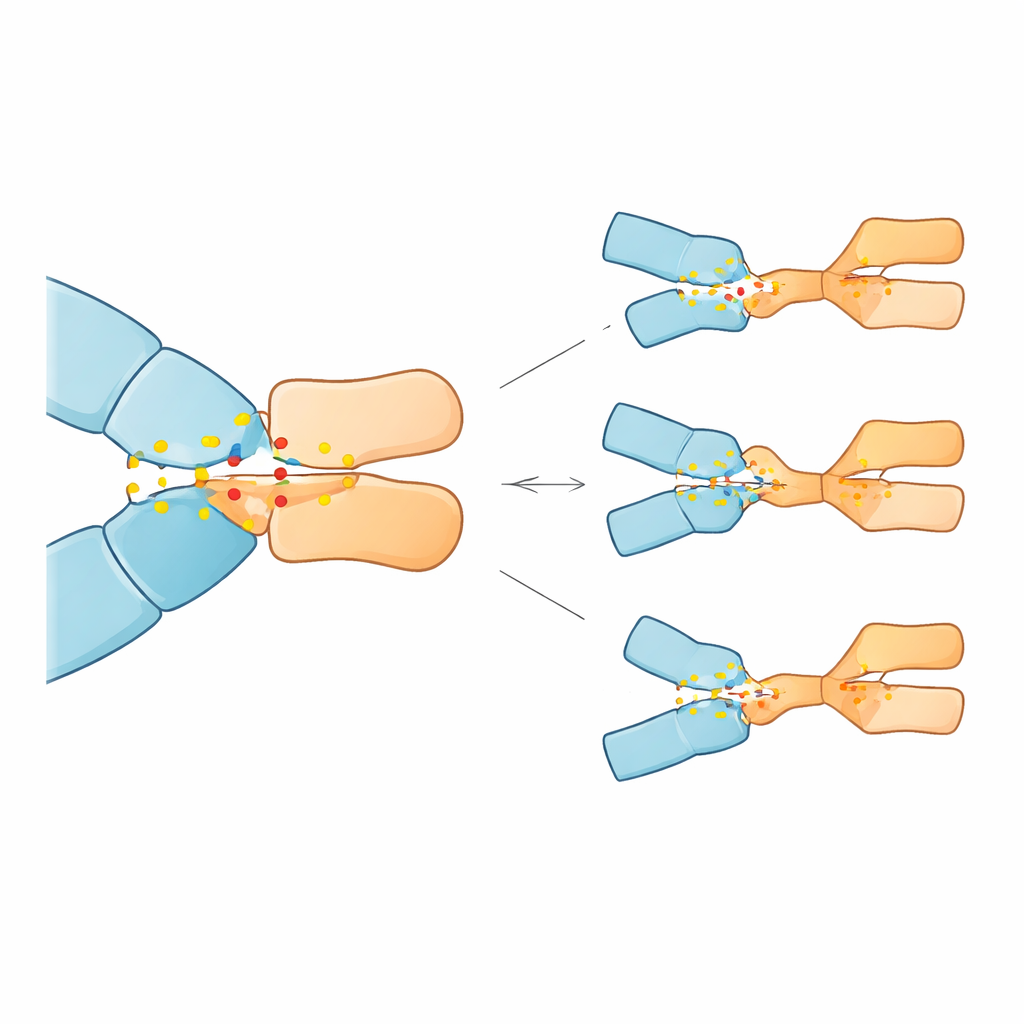
Wanneer belangrijke ankers verloren gaan
De simulaties toonden aan dat 23 van de 28 mutaties de pembrolizumab-binding verzwakten, sommige behoorlijk dramatisch, terwijl vijf deze juist versterkten. Drie mutaties vielen op door het veroorzaken van het grootste voorspelde verlies aan affiniteit: substituties op posities 85 en 89 van PD-1 (Asp85Gly, Asp85Asn en Pro89Arg). In het normale eiwit fungeert Asp85 als een elektrostatief anker en vormt het een robuuste zoutbrug en waterstofbruggen met een arginine in het antilichaam; Pro89 helpt bij het vormen van een cluster van nauw aansluitende hydrofobe contacten met meerdere antilichaamresiduen. De Asp85-mutaties verwijderden de negatieve lading of het vermogen tot waterstofbinding op deze plaats, waardoor het anker effectief werd verbroken en het omliggende netwerk van contacten werd gedestabiliseerd. De Pro89Arg-wijziging verving een neutraal, vormpassend residu door een positief geladen residu, waardoor de hydrofobe cluster werd verstoord en zijn stabiliserende bijdrage afnam. Daarentegen hervormden enkele andere mutaties op verschillende posities het interface subtiel op manieren die de binding konden verbeteren, althans in het model.
Wat dit voor patiënten zou kunnen betekenen
Deze bevindingen suggereren dat zelfs zeer zeldzame veranderingen in het PD-1-gen, als ze optreden op cruciale contactpunten, kunnen verminderen hoe goed pembrolizumab zich vasthecht aan zijn doel en daardoor het vermogen om T-cellen tegen kanker los te maken kunnen afremmen. Hoewel het werk volledig computationeel is en experimentele en klinische bevestiging nodig heeft, biedt het een routekaart: specifieke PD-1-varianten zoals die op posities 85 en 89 zouden ooit als biomarkers kunnen dienen om patiënten met een hoger risico op resistentie te signaleren of om de keuze tussen verschillende PD-1-gerichte geneesmiddelen te begeleiden. Breder gezien illustreert de studie hoe diepgaande computersimulaties de fijnmazige moleculaire redenen kunnen verhelderen waarom krachtige immunotherapieën bij sommige mensen slagen maar bij anderen falen.
Bronvermelding: Bouricha, E.M., Hakmi, M., Batlamous, B. et al. The impact of PD-1 mutations on pembrolizumab binding: insights from molecular dynamics and MM-GBSA analysis. Sci Rep 16, 10773 (2026). https://doi.org/10.1038/s41598-026-45077-0
Trefwoorden: PD-1-mutaties, pembrolizumab-resistentie, kankerimmunotherapie, remmers van immuuncheckpoints, moleculaire dynamica-simulatie