Clear Sky Science · pl
Przewidywany przez uczenie maszynowe krajobraz organizacji chromatyny w nowotworach pediatrycznych
Dlaczego fałdowanie genomu ma znaczenie dla chorych dzieci
Nowotwory mózgu u dzieci należą do najgroźniejszych nowotworów wieku dziecięcego, a w wielu przypadkach lekarze wciąż nie wiedzą dokładnie, co poszło nie tak w ich DNA. W badaniu tym zbadano nowe ujęcie problemu: nie tylko które geny są zmutowane, lecz także w jaki sposób duże przestawienia DNA zmieniają sposób, w jaki genom składa się wewnątrz komórki. Łącząc obszerne zbiory danych onkologicznych z potężnym modelem uczenia maszynowego, autorzy pokazują, że ukryte zaburzenia tego trójwymiarowego wzorca fałdowania mogą napędzać nowotwory dziecięce — i że komputery potrafią już wytypować najbardziej niebezpieczne zmiany do dalszych badań.
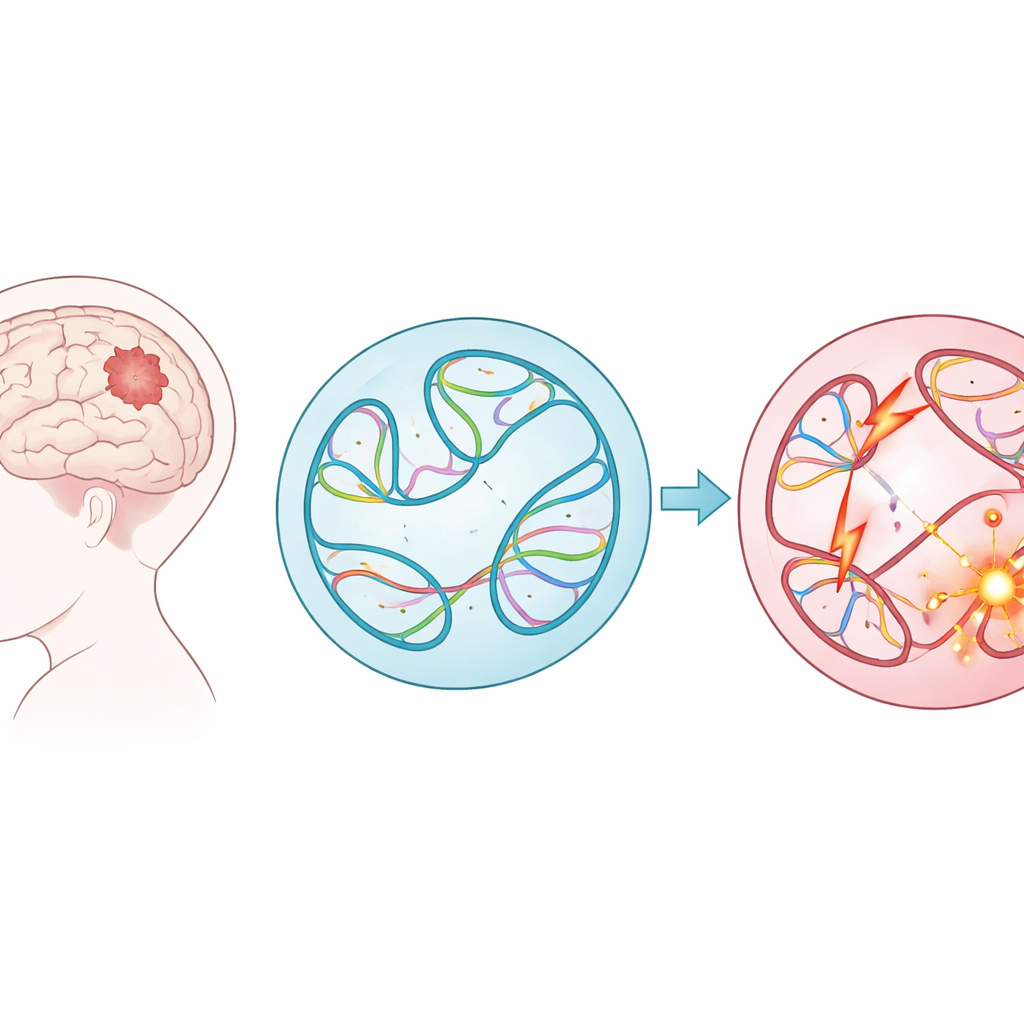
Duże przetasowania DNA w nowotworach dziecięcych
Zamiast koncentrować się na drobnych literówkach w kodzie genetycznym, badacze przyjrzeli się wariantom strukturalnym — dużym wycięciom, duplikacjom, odwróceniom i wstawkom długich odcinków DNA. Takie zmiany na dużą skalę mogą zlepić geny, usuwać regiony ochronne albo umieszczać silne przełączniki (enhancery) obok niewłaściwych genów. Wykorzystując dane z prawie 1 900 dzieci z Children’s Brain Tumor Network, obejmujące 61 typów nowotworów, zespół skatalogował niemal 300 000 takich wariantów. Stwierdzili, że niektóre grupy nowotworów, takie jak chłoniaki i mięsaki, miały znacznie więcej tych zmian niż inne, a guzy nawrotowe lub progresywne po początkowym leczeniu zwykle zawierały więcej wariantów strukturalnych niż guzy pierwotne.
Wykorzystanie AI do zobaczenia genomu w 3D
Bezpośrednie zmierzenie, jak każdy wariant strukturalny zmienia fałdowanie DNA w komórce, wymagałoby pracochłonnych eksperymentów dla setek tysięcy miejsc — praktycznie niemożliwe w tej skali. Zamiast tego autorzy wykorzystali sieć konwolucyjną o nazwie Akita, udostępnioną przez ich pipeline SuPreMo-Akita, aby przewidzieć, jak milionparzaznacznikowy fragment DNA fałduje się w 3D. Dla każdego wariantu strukturalnego symulowali lokalną sekwencję DNA z wprowadzoną i bez wprowadzanej zmiany, poprosili model o przewidzenie map kontaktów — wzorców pokazujących, które części genomu się stykają — i porównali te mapy. Im większa różnica, tym silniejsze przewidywane zaburzenie organizacji genomu przez dany wariant. Pozwoliło to na uszeregowanie wariantów we wszystkich guzach według przewidywanej siły ich wpływu na normalne wzorce fałdowania.
Miejsca zapalne, gdzie fałdowanie zawodzi
Przeszukując genom pod kątem miejsc wielokrotnie dotykanych przez silnie zaburzające warianty, zespół odkrył pięć nawracająco zakłóconych regionów — odcinków DNA, gdzie guzy wielu dzieci, różnych typów, wykazywały silne przewidywane uszkodzenia lokalnego fałdowania. W kilku z tych regionów model wskazywał utratę kluczowych cech strukturalnych, takich jak granice domen czy pętle, które normalnie separują grupy genów i ich przełączników. Co niezwykłe, niektóre z tych hotspotów nie były szczególnie silnie zmutowane pod względem liczby zmian; wyróżniała je natomiast dotkliwość zaburzenia fałdowania, gdy warianty występowały właśnie tam. Regiony te zawierały geny zaangażowane w rozwój mózgu i znane funkcje związane z nowotworami, sugerując, że subtelne trójwymiarowe błędy połączeń, a nie sama liczba mutacji, mogą mieć największe znaczenie.
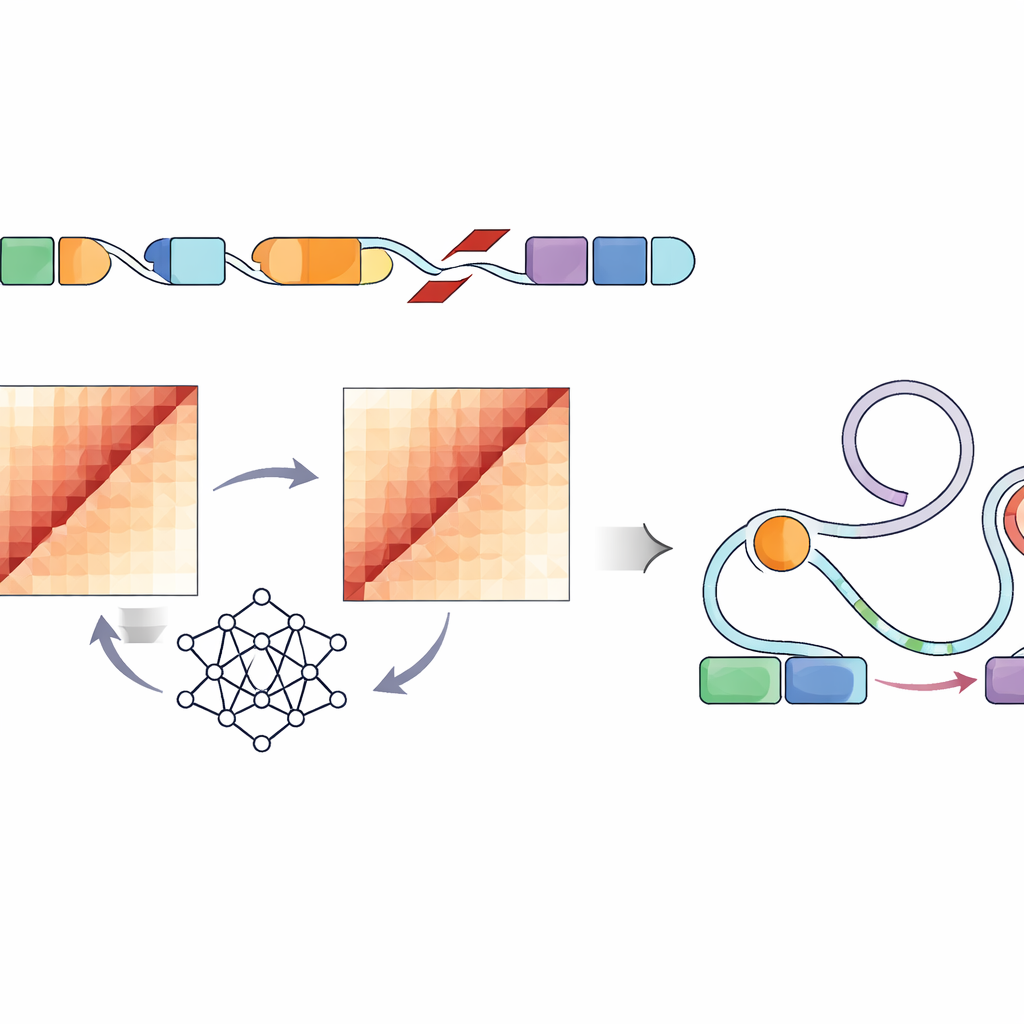
Łączenie zaburzeń fałdowania z przełącznikami kontroli genów
Ponieważ nie każda zmiana kształtu DNA wpływa na zachowanie komórek, badacze przyjrzeli się następnie elementom regulacyjnym — enhancerom oznaczonym charakterystycznymi znakami chemicznymi i otwartą chromatyną w liniach komórkowych przypominających guz. Odkryli, że silnie zaburzające warianty w kilku typach nowotworów pediatrycznych częściej lokalizowały się w tych aktywnych regionach enhancerowych lub w ich pobliżu. Opierając się na istniejącym modelu Activity-by-Contact, stworzyli „wynik zaburzenia ABC”, który podbija wagę wariantów przewidywanych do zakłócania fałdowania genomu szczególnie tam, gdzie znajdują się te enhancery. To dopracowane ocenianie odsłoniło warianty strukturalne, które silnie zmieniały przewidywane kontakty między enhancerami a pobliskimi genami związanymi z wzrostem komórek, przeżyciem i funkcjami mózgu, w tym dobrze znanymi genami nowotworowymi takimi jak PDGFRA, ID2, MYCN i innymi.
Wskazówki do nowych czynników napędzających raka i przyszłej opieki
Skupiając się na szczególnie agresywnym typie guza zwanym atypical teratoid/rhabdoid tumor, metoda wyróżniła przestawienia w pobliżu genów zaangażowanych w remodelowanie chromatyny, naprawę DNA i rozwój neuronów. W kilku przypadkach guzy noszące te warianty wykazywały także wyjątkowo wysoką lub niską ekspresję pobliskich genów, co zgadza się z mechanizmem „przejęcia” enhancerów przez zmienione kontakty 3D. Chociaż te obserwacje wymagają jeszcze potwierdzenia eksperymentalnego, wskazują na potężny nowy sposób przesiewania ogromnej liczby wariantów strukturalnych i priorytetyzowania tych, które najprawdopodobniej wpływają na zachowanie guza. W dłuższej perspektywie mapy krajobrazu fałdowania genomu kierowane przez uczenie maszynowe mogą pomóc lekarzom interpretować wyniki sekwencjonowania u małych pacjentów z nowotworami, odkrywać ukrytych czynników choroby i ostatecznie ukierunkować poszukiwania bardziej precyzyjnych, mniej toksycznych terapii.
Cytowanie: Gjoni, K., Zhang, S., Yan, R.E. et al. Machine learning-predicted chromatin organization landscape across pediatric tumors. Sci Rep 16, 10790 (2026). https://doi.org/10.1038/s41598-026-44925-3
Słowa kluczowe: nowotwory mózgu u dzieci, warianty strukturalne, 3D organizacja genomu, genomika i uczenie maszynowe, przejęcie aktywatorów (enhancer hijacking)