Clear Sky Science · it
Paesaggio dell’organizzazione della cromatina previsto dall’apprendimento automatico nei tumori pediatrici
Perché il ripiegamento del genoma conta per i bambini malati
I tumori cerebrali pediatrici sono tra i tumori infantili più letali, eppure per molti giovani pazienti i medici non sanno ancora esattamente cosa sia andato storto nel loro DNA. Questo studio esplora una nuova prospettiva: non solo quali geni sono mutati, ma come le grandi riorganizzazioni del DNA modificano il modo in cui il genoma si ripiega all’interno di ogni cellula. Combinando ampi dataset tumorali con un potente modello di apprendimento automatico, gli autori mostrano che interruzioni nascoste in questo schema di ripiegamento 3D possono contribuire allo sviluppo dei tumori pediatrici — e che i computer possono ora individuare i cambiamenti più pericolosi per studi successivi.
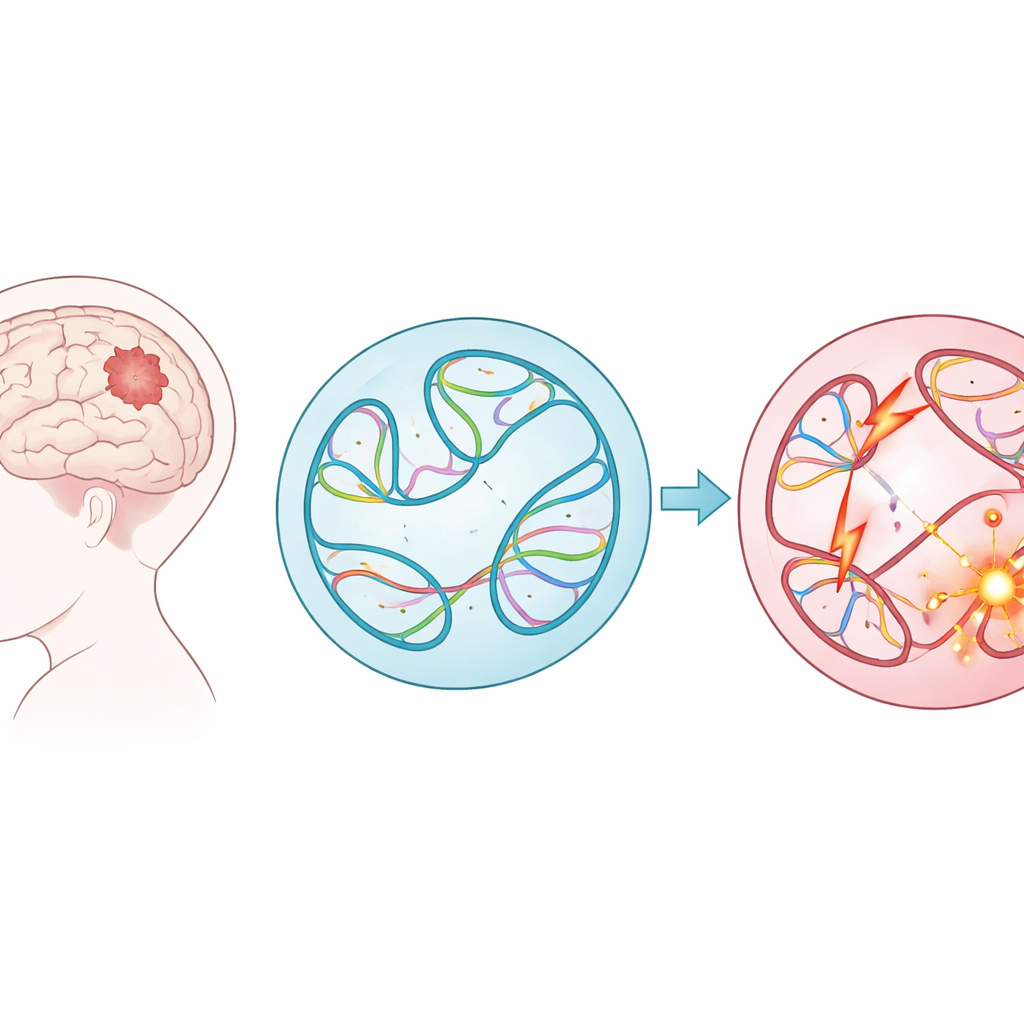
Grandi rimescolamenti di DNA nei tumori infantili
Invece di concentrarsi su piccoli errori nella sequenza genetica, i ricercatori hanno esaminato le varianti strutturali — grandi tagli, duplicazioni, inversioni e incollaggi di estesi tratti di DNA. Questi cambiamenti su larga scala possono fondere geni, eliminare regioni protettive o porre potenti interruttori (potenziatori) accanto ai geni sbagliati. Utilizzando dati di quasi 1.900 bambini nella Children’s Brain Tumor Network, che coprono 61 tipi di tumore, il team ha catalogato quasi 300.000 di queste varianti. Hanno scoperto che alcuni gruppi tumorali, come i linfomi e i sarcomi, presentavano molte più di queste alterazioni rispetto ad altri, e che i tumori recidivati o progrediti dopo il trattamento iniziale tipicamente ospitavano più varianti strutturali rispetto ai tumori originali.
Usare l’IA per vedere il genoma in 3D
Misurare direttamente come ogni variante strutturale modifica il ripiegamento del DNA all’interno di una cellula richiederebbe esperimenti laboriosi per centinaia di migliaia di siti — sostanzialmente impossibile su questa scala. Invece, gli autori hanno utilizzato una rete neurale convoluzionale chiamata Akita, accessibile tramite la loro pipeline SuPreMo-Akita, per prevedere come si ripiega in 3D un tratto di DNA di un milione di basi. Per ogni variante strutturale hanno simulato la sequenza locale con e senza la modifica, chiesto al modello di prevedere mappe di contatto — schemi che mostrano quali parti del genoma si toccano — e poi confrontato queste mappe. Maggiore è la differenza, maggiore è la previsione che quella variante disturbi l’organizzazione del genoma. Questo ha permesso loro di classificare le varianti attraverso tutti i tumori in base alla forza con cui si pensava alterassero o spezzassero i normali schemi di ripiegamento.
Punti caldi in cui il ripiegamento va storto
Quando il team ha scandagliato il genoma per trovare luoghi ripetutamente colpiti da varianti altamente distruttive, ha scoperto cinque regioni ricorrentemente danneggiate — tratti di DNA in cui i tumori di molti bambini, di vari tipi, mostravano un forte danno previsto al ripiegamento locale. In diverse di queste regioni il modello ha indicato la perdita di caratteristiche strutturali chiave, come confini di dominio e loop che normalmente separano gruppi di geni e i loro interruttori. Colpisce il fatto che alcune di queste aree non fossero particolarmente mutate in termini assoluti; ciò che le distingueva era la gravità della perturbazione del ripiegamento quando le varianti si verificavano lì. Queste regioni contenevano geni coinvolti nello sviluppo cerebrale e in funzioni correlate al cancro, suggerendo che un malfunzionamento 3D sottile, più che il semplice conteggio di mutazioni, potrebbe essere ciò che conta davvero.
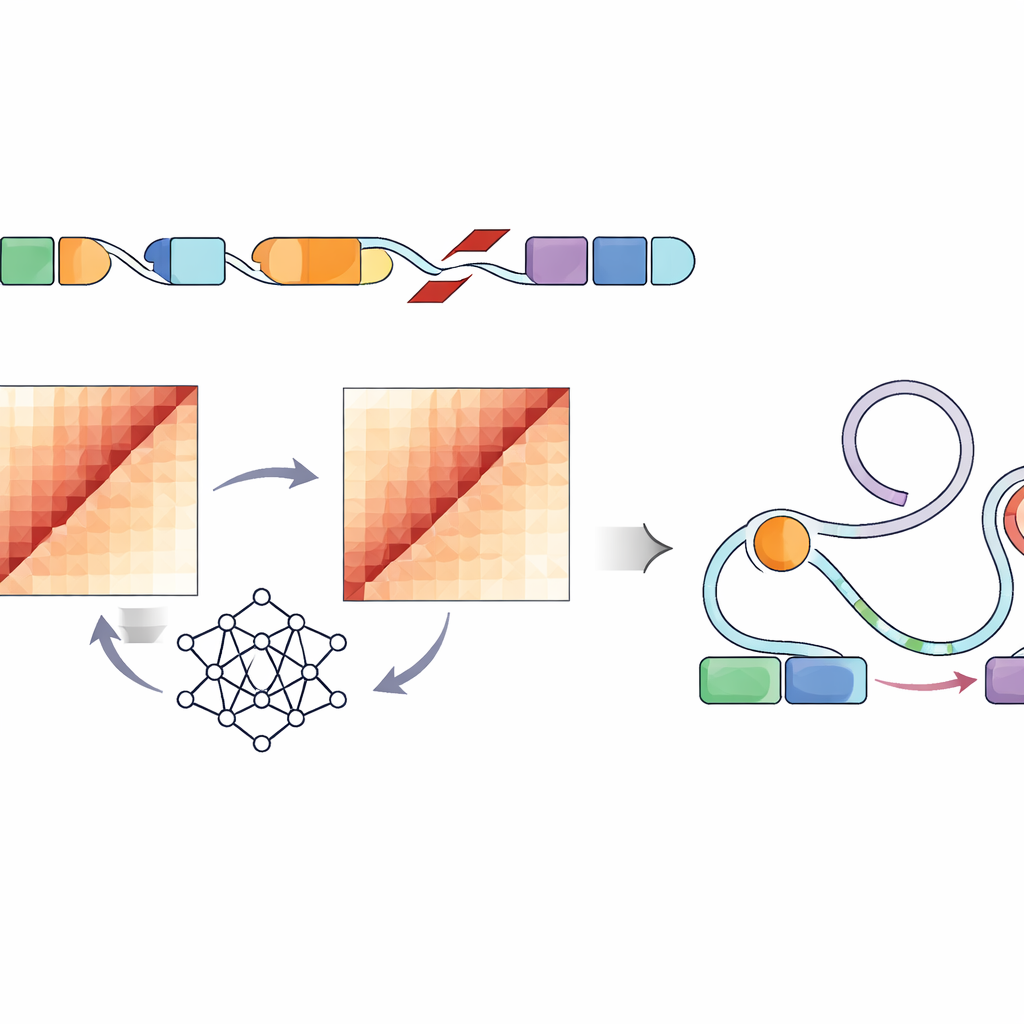
Collegare le perturbazioni del ripiegamento agli interruttori di controllo genico
Poiché non tutte le modifiche della forma del DNA influiscono sul comportamento cellulare, i ricercatori hanno poi esaminato specificamente gli elementi regolatori — potenziatori contrassegnati da particolari marcatori chimici e da cromatina aperta in linee cellulari simili a tumori. Hanno rilevato che le varianti altamente distruttive in diversi tipi di tumore pediatrico tendevano a trovarsi dentro o vicino a queste regioni di potenziatori attivi. Basandosi su un quadro esistente chiamato modello Activity-by-Contact, hanno creato un “punteggio di interruzione ABC” che valorizza le varianti previste disturbare il ripiegamento del genoma proprio dove risiedono questi potenziatori. Questa classificazione raffinata ha messo in luce varianti strutturali che modificavano fortemente i contatti previsti tra potenziatori e geni vicini con ruoli nella crescita cellulare, nella sopravvivenza e nella funzione cerebrale, inclusi geni noti coinvolti nel cancro come PDGFRA, ID2, MYCN e altri.
Indizi per nuovi driver del cancro e per le cure future
Concentrandosi su un tipo di tumore particolarmente aggressivo chiamato tumore teratoide/rabdoide atipico, il metodo ha evidenziato riorganizzazioni vicino a geni coinvolti nel rimodellamento della cromatina, nella riparazione del DNA e nello sviluppo neuronale. In diversi casi, i tumori che portavano queste varianti mostravano anche espressioni insolitamente alte o basse dei geni vicini, coerente con il “sequestro” di potenziatori tramite contatti 3D alterati. Pur richiedendo ancora conferme sperimentali, questi risultati indicano un nuovo modo potente per setacciare enormi quantità di varianti strutturali e dare priorità a quelle più probabilmente influenti sul comportamento tumorale. A lungo termine, mappe guidate dall’apprendimento automatico del paesaggio del ripiegamento del genoma potrebbero aiutare i medici a interpretare i risultati del sequenziamento nei giovani pazienti oncologici, scoprire driver nascosti della malattia e, in ultima analisi, orientare la ricerca verso terapie più precise e meno tossiche.
Citazione: Gjoni, K., Zhang, S., Yan, R.E. et al. Machine learning-predicted chromatin organization landscape across pediatric tumors. Sci Rep 16, 10790 (2026). https://doi.org/10.1038/s41598-026-44925-3
Parole chiave: tumori cerebrali pediatrici, varianti strutturali, organizzazione 3D del genoma, genomica con apprendimento automatico, sequestro di potenziatori