Clear Sky Science · nl
Door machine learning voorspelde landschap van chromatineorganisatie in pediatrische tumoren
Waarom de vouwing van het genoom ertoe doet bij zieke kinderen
Pediatrische hersentumoren behoren tot de dodelijkste kinderkankers, maar voor veel jonge patiënten weten artsen nog steeds niet precies wat er in hun DNA misging. Deze studie verkent een nieuw perspectief: niet alleen welke genen gemuteerd zijn, maar hoe grote DNA-herschikkingen de manier veranderen waarop het genoom in elke cel opvouwt. Door grote kanker‑datasets te combineren met een krachtig machine‑learningmodel laten de auteurs zien dat verborgen verstoringen in dit 3D‑vouwwerk kunnen bijdragen aan kindertumoren — en dat computers nu de meest gevaarlijke veranderingen kunnen signaleren voor nader onderzoek.
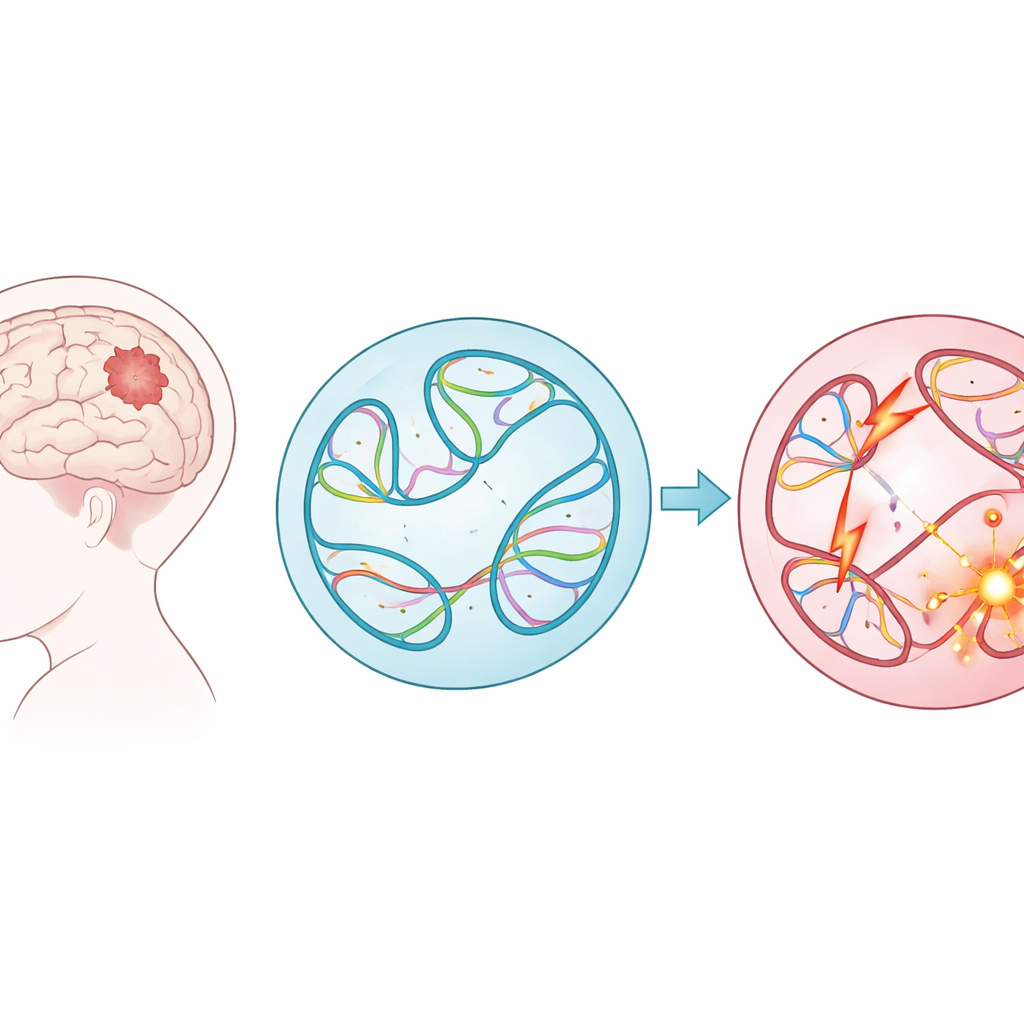
Grote DNA‑verschuivingen in kindertumoren
In plaats van zich te richten op kleine foutjes in de genetische code onderzochten de onderzoekers structurele varianten — grote knip-, kopieer-, omkeer- en plakbewerkingen van lange DNA‑stukken. Deze grootschalige veranderingen kunnen genen aan elkaar plakken, beschermende regio’s verwijderen of krachtige schakelaars (enhancers) naast de verkeerde genen plaatsen. Met gegevens van bijna 1.900 kinderen uit het Children’s Brain Tumor Network, verspreid over 61 tumortypen, bracht het team bijna 300.000 dergelijke varianten in kaart. Ze vonden dat sommige tumorgroepen, zoals lymfomen en sarcomen, veel meer van deze afwijkingen droegen dan andere, en dat tumoren die terugkeerden of vooruitgingen na de eerste behandeling doorgaans meer structurele varianten bevatten dan de oorspronkelijke tumoren.
AI gebruiken om het genoom in 3D te zien
Direct meten hoe elke structurele variant de vouwing van DNA in een cel verandert, zou arbeidsintensieve experimenten voor honderdduizenden sites vereisen — praktisch onmogelijk op deze schaal. In plaats daarvan gebruikten de auteurs een convolutioneel neuraal netwerk genaamd Akita, toegankelijk via hun SuPreMo‑Akita‑pipeline, om te voorspellen hoe een stuk DNA van één miljoen baseparen in 3D vouwt. Voor elke structurele variant simuleerden ze de lokale DNA‑sequentie met en zonder de wijziging, lieten het model contactkaarten voorspellen — patronen die laten zien welke delen van het genoom elkaar raken — en vergeleken die kaarten. Hoe groter het verschil, hoe meer die variant werd voorspeld de genoomorganisatie te verstoren. Dit stelde hen in staat varianten over alle tumoren te rangschikken op basis van hoe sterk ze naar verwachting normale vouwwerkpatronen zouden buigen of breken.
Warmtepunten waar vouwing fout gaat
Toen het team het genoom doorspeurde op plaatsen die herhaaldelijk werden getroffen door sterk verstorende varianten, ontdekten ze vijf herhaaldelijk verstoorde regio’s — DNA‑streken waar in veel kindertumoren van verschillende typen sterke voorspelde schade aan de lokale vouwing voorkwam. In meerdere van deze regio’s gaf het model aan dat sleutelstructuren zoals domeingrenzen en lussen verloren gingen, die normaal groepen genen en hun schakelaars scheiden. Opvallend was dat sommige van deze hotspots niet per se veel mutaties in totaal hadden; wat hen onderscheidde was de ernst van de vouwingverstoring wanneer varianten daar optraden. Deze regio’s bevatten genen die betrokken zijn bij hersenontwikkeling en bekende kankergerelateerde functies, wat suggereert dat subtiele 3D‑verkeerdbedrading, in plaats van alleen mutatieaantallen, het belangrijkste kan zijn.
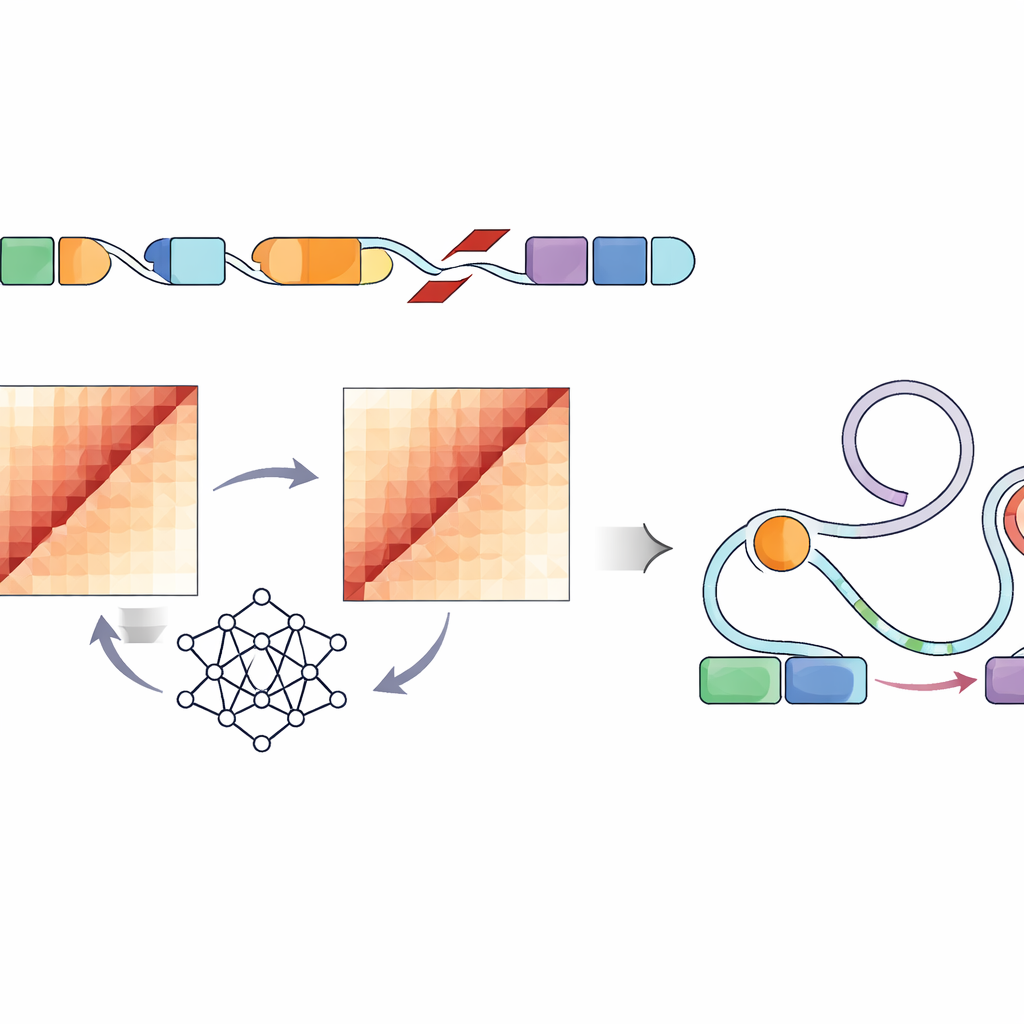
Het koppelen van vouwingverstoring aan genregulatieschakelaars
Aangezien niet elke verandering in DNA‑vorm de celgedrag zal beïnvloeden, bekeken de onderzoekers vervolgens specifiek regulatorische elementen — enhancers gemarkeerd door karakteristieke chemische tags en open chromatine in tumorachtige cellijnen. Ze vonden dat sterk verstorende varianten in meerdere pediatrische tumortypen vaker in of nabij deze actieve enhancerregio’s lagen. Uitgaand van een bestaand kader, het Activity‑by‑Contact‑model, maakten ze een “ABC‑disruptiescore” die varianten bevoordeelt waarvan wordt voorspeld dat ze de genoomvouwing verstoren precies waar deze enhancers zich bevinden. Deze verfijnde score bracht structurele varianten naar voren die naar verwachting sterk de voorspelde contacten tussen enhancers en nabijgelegen genen veranderden met rollen in celgroei, overleving en hersenfunctie, waaronder bekende kankergenes zoals PDGFRA, ID2, MYCN en anderen.
Aanwijzingen voor nieuwe kankerdrijvers en toekomstige zorg
Met een focus op een bijzonder agressief tumortype, het atypische teratoïde/rhabdoïde tumor, wees de methode herschikkingen aan nabij genen die betrokken zijn bij chromatineremodellering, DNA‑herstel en neurale ontwikkeling. In verschillende gevallen lieten de tumoren met deze varianten ook ongewoon hoge of lage expressie van de nabijgelegen genen zien, wat consistent is met enhancer “hijacking” via veranderde 3D‑contacten. Hoewel deze bevindingen nog experimentele bevestiging vereisen, wijzen ze op een krachtige nieuwe manier om enorme aantallen structurele varianten te doorzoeken en die te prioriteren die het meest waarschijnlijk het tumorgedrag beïnvloeden. Op de lange termijn zouden dergelijke door machine learning geleide kaarten van het vouwnlandschap van het genoom artsen kunnen helpen sequentieresultaten van jonge kankerpatiënten te interpreteren, verborgen drijvers van ziekte te onthullen en uiteindelijk de zoektocht naar meer precieze, minder toxische behandelingen te sturen.
Bronvermelding: Gjoni, K., Zhang, S., Yan, R.E. et al. Machine learning-predicted chromatin organization landscape across pediatric tumors. Sci Rep 16, 10790 (2026). https://doi.org/10.1038/s41598-026-44925-3
Trefwoorden: pediatrische hersentumoren, structurele varianten, 3D-genoomorganisatie, machine learning genomica, enhancer hijacking