Clear Sky Science · es
Paisaje de la organización de la cromatina predicho por aprendizaje automático en tumores pediátricos
Por qué importa el plegamiento del genoma en niños enfermos
Los tumores cerebrales pediátricos están entre los cánceres infantiles más letales, y sin embargo en muchos pacientes jóvenes los médicos aún no saben con precisión qué falló en su ADN. Este estudio aborda un ángulo nuevo: no solo qué genes están mutados, sino cómo las grandes reordenaciones del ADN cambian la forma en que el genoma se pliega dentro de cada célula. Al combinar grandes conjuntos de datos sobre cáncer con un potente modelo de aprendizaje automático, los autores muestran que las alteraciones ocultas en este patrón de plegamiento 3D pueden contribuir al desarrollo de tumores infantiles, y que los ordenadores ya pueden señalar los cambios más peligrosos para un análisis posterior.
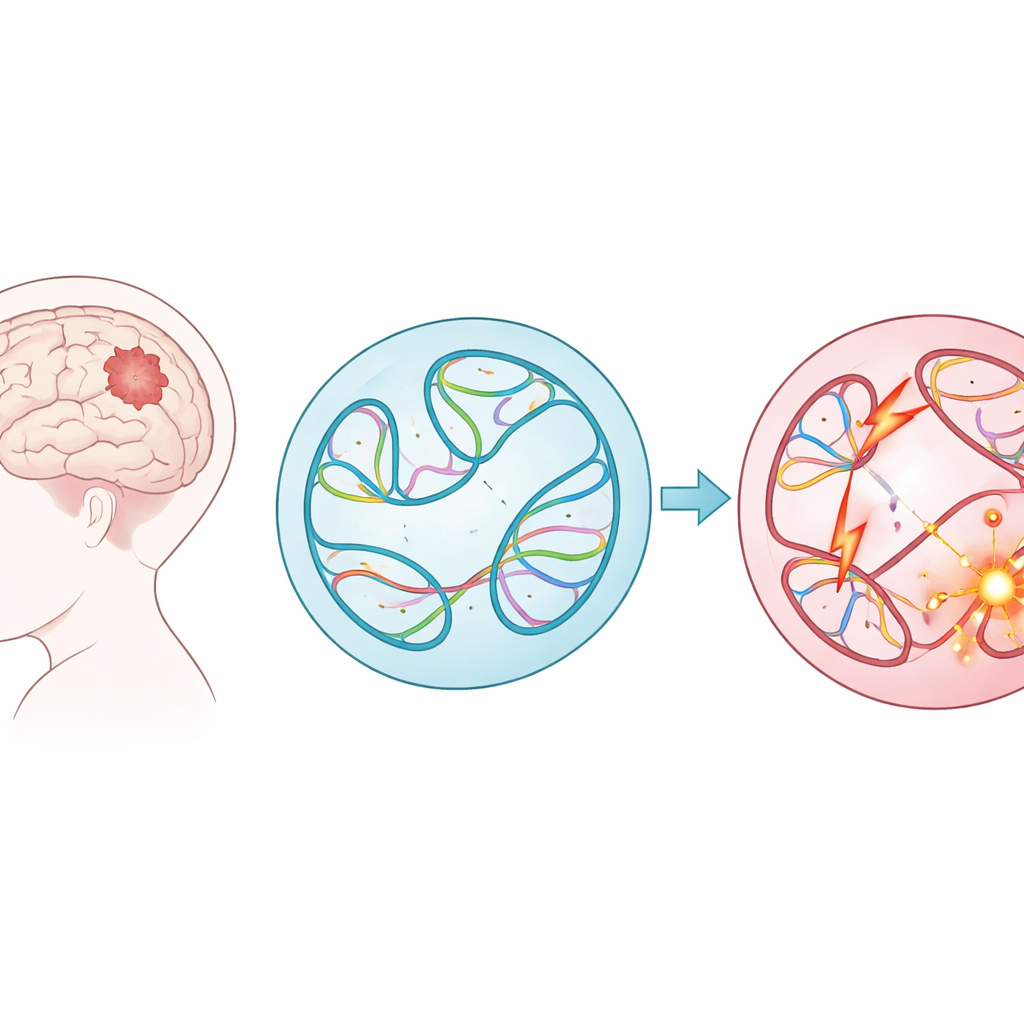
Grandes remezclas del ADN en tumores infantiles
En lugar de centrarse en pequeños errores tipográficos en el código genético, los investigadores examinaron variantes estructurales: grandes cortes, duplicaciones, inversiones y transposiciones de largos tramos de ADN. Estos cambios a gran escala pueden fusionar genes, eliminar regiones protectoras o situar interruptores potentes (potenciadores) junto a genes equivocados. Con datos de casi 1.900 niños de la Children’s Brain Tumor Network, que abarcan 61 tipos de tumor, el equipo catalogó casi 300.000 de estas variantes. Encontraron que algunos grupos de tumores, como linfomas y sarcomas, presentaban muchas más de estas alteraciones que otros, y que los tumores recurrentes o en progresión tras el tratamiento inicial solían albergar más variantes estructurales que los tumores originales.
Usar IA para ver el genoma en 3D
Medir directamente cómo cada variante estructural cambia el plegamiento del ADN dentro de una célula exigiría experimentos laboriosos para cientos de miles de sitios—prácticamente imposible a esta escala. En su lugar, los autores emplearon una red neuronal convolucional llamada Akita, accesible a través de su canalización SuPreMo-Akita, para predecir cómo se pliega en 3D un tramo de ADN de un millón de pares de bases. Para cada variante estructural, simularon la secuencia local de ADN con y sin la alteración, pidieron al modelo que predijera mapas de contacto—patrones que muestran qué partes del genoma se tocan—y luego compararon estos mapas. Cuanto mayor era la diferencia, más se predecía que esa variante perturbaba la organización genómica. Esto les permitió clasificar las variantes en todos los tumores según la intensidad con la que se esperaba que doblaran o rompieran los patrones normales de plegamiento.
Puntos críticos donde el plegamiento falla
Al rastrear el genoma en busca de lugares golpeados repetidamente por variantes altamente disruptivas, identificaron cinco regiones recurrentemente dañadas—tramos de ADN donde los tumores de muchos niños, de distintos tipos, mostraban un daño local al plegamiento predicho con gran intensidad. En varias de estas regiones, el modelo indicó pérdida de características estructurales clave, como límites de dominio y bucles que normalmente separan grupos de genes y sus interruptores. De forma llamativa, algunos de estos puntos críticos no estaban especialmente mutados en términos de recuento total; lo que los distinguía era la severidad de la alteración del plegamiento cuando aparecían variantes allí. Estas regiones contenían genes implicados en el desarrollo cerebral y en funciones relacionadas con el cáncer, lo que sugiere que un cableado 3D sutil mal dirigido, más que el simple recuento de mutaciones, puede ser lo que realmente importa.
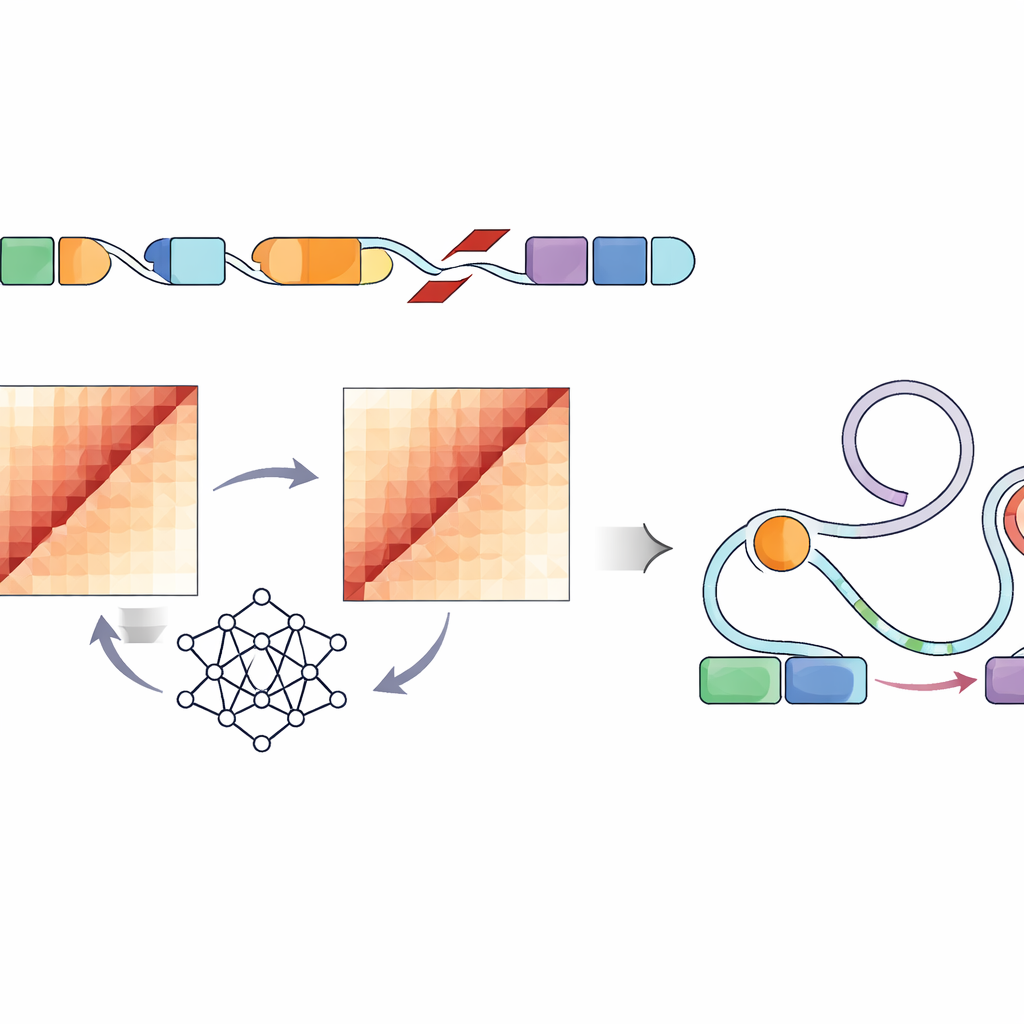
Vinculando las alteraciones del plegamiento con los interruptores reguladores
Como no todos los cambios en la forma del ADN afectarán al comportamiento celular, los investigadores examinaron a continuación elementos regulatorios específicos—potenciadores marcados por etiquetas químicas características y cromatina abierta en líneas celulares similares a tumores. Encontraron que las variantes altamente disruptivas en varios tipos de tumor pediátrico tendían a ubicarse en o cerca de estas regiones activas de potenciadores. Basándose en un marco existente llamado Activity-by-Contact, crearon una “puntuación de disrupción ABC” que pondera las variantes previstas para perturbar el plegamiento genómico específicamente donde residen estos potenciadores. Esta puntuación refinada sacó a la luz variantes estructurales que alteraban fuertemente los contactos previstos entre potenciadores y genes cercanos con papeles en el crecimiento celular, la supervivencia y la función cerebral, incluidos genes oncológicos bien conocidos como PDGFRA, ID2, MYCN y otros.
Pistas sobre nuevos conductores del cáncer y la atención futura
Al centrarse en un tipo de tumor particularmente agresivo llamado tumor teratoide/rabdóide atípico, el método destacó reordenamientos cerca de genes involucrados en la remodelación de la cromatina, la reparación del ADN y el desarrollo neuronal. En varios casos, los tumores portadores de estas variantes también mostraron una expresión inusualmente alta o baja de los genes cercanos, consistente con el “secuestro” de potenciadores mediante contactos 3D alterados. Aunque estos hallazgos aún requieren confirmación experimental, apuntan a una manera potente de cribar enormes cantidades de variantes estructurales y priorizar aquellas con más probabilidad de influir en el comportamiento tumoral. A largo plazo, mapas del paisaje de plegamiento del genoma guiados por aprendizaje automático podrían ayudar a los médicos a interpretar resultados de secuenciación en pacientes pediátricos con cáncer, descubrir conductores ocultos de la enfermedad y, en última instancia, orientar la búsqueda de tratamientos más precisos y menos tóxicos.
Cita: Gjoni, K., Zhang, S., Yan, R.E. et al. Machine learning-predicted chromatin organization landscape across pediatric tumors. Sci Rep 16, 10790 (2026). https://doi.org/10.1038/s41598-026-44925-3
Palabras clave: tumores cerebrales pediátricos, variantes estructurales, organización 3D del genoma, genómica con aprendizaje automático, secuestro de potenciadores