Clear Sky Science · en
Machine learning-predicted chromatin organization landscape across pediatric tumors
Why the Genome’s Folding Matters for Sick Kids
Pediatric brain tumors are among the deadliest childhood cancers, yet for many young patients doctors still do not know exactly what went wrong in their DNA. This study explores a new angle: not just which genes are mutated, but how large DNA rearrangements change the way the genome folds inside each cell. By combining big cancer datasets with a powerful machine-learning model, the authors show that hidden disruptions in this 3D folding pattern may help drive childhood tumors—and that computers can now flag the most dangerous changes for further study.
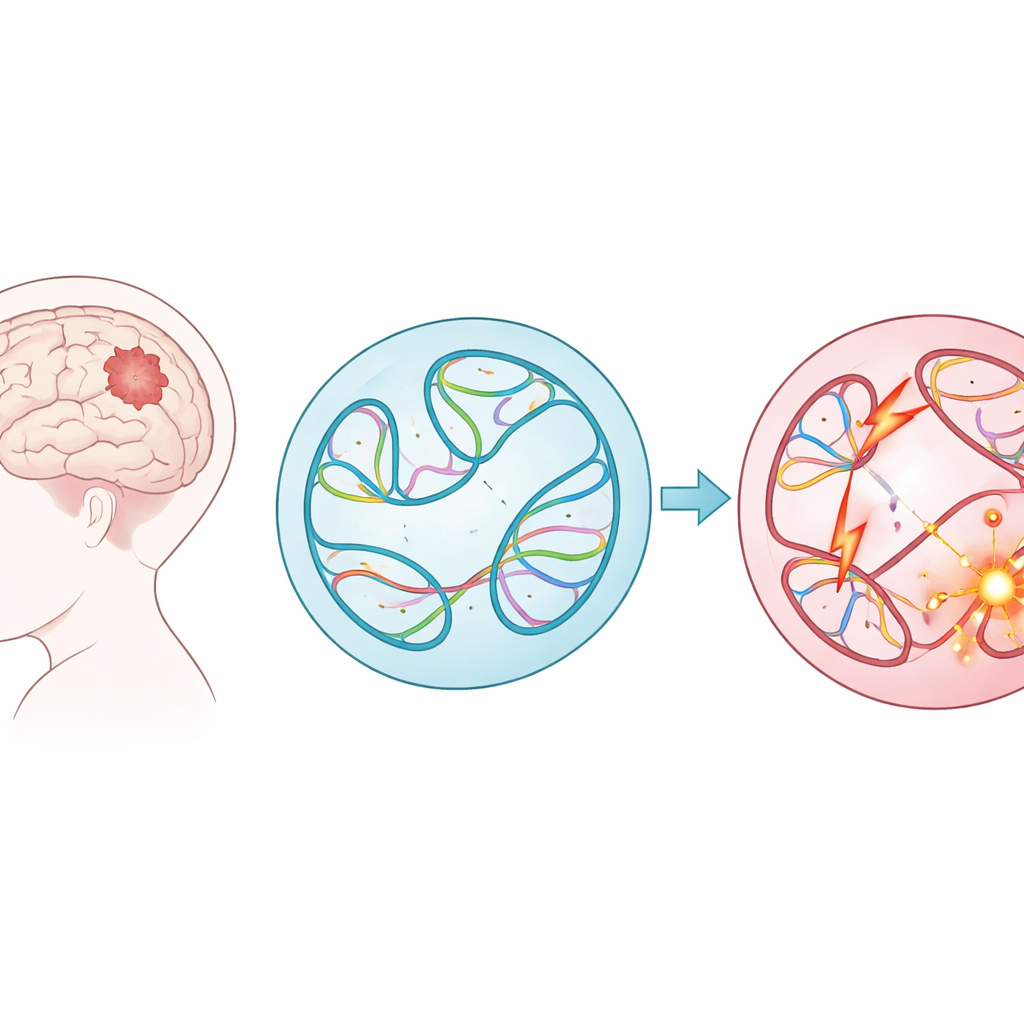
Big DNA Shuffles in Childhood Tumors
Instead of focusing on small typos in the genetic code, the researchers examined structural variants—big cuts, copies, flips, and pastes of long stretches of DNA. These large-scale changes can fuse genes together, delete protective regions, or place powerful switches (enhancers) next to the wrong genes. Using data from nearly 1,900 children in the Children’s Brain Tumor Network, spanning 61 tumor types, the team cataloged almost 300,000 such variants. They found that some tumor groups, such as lymphomas and sarcomas, carried many more of these alterations than others, and that tumors that had come back or progressed after initial treatment typically harbored more structural variants than the original tumors.
Using AI to See the Genome in 3D
Directly measuring how each structural variant changes the folding of DNA inside a cell would require laborious experiments for hundreds of thousands of sites—essentially impossible at this scale. Instead, the authors used a convolutional neural network called Akita, accessed through their SuPreMo-Akita pipeline, to predict how a one-million–base-pair stretch of DNA folds in 3D. For every structural variant, they simulated the local DNA sequence with and without the change, asked the model to predict contact maps—patterns showing which parts of the genome touch—and then compared these maps. The bigger the difference, the more that variant was predicted to disturb genome organization. This let them rank variants across all tumors by how strongly they were expected to bend or break normal folding patterns.
Hotspots Where Folding Goes Wrong
When the team scanned the genome for places repeatedly hit by highly disruptive variants, they uncovered five recurrently disrupted regions—stretches of DNA where many children’s tumors, of various types, showed strong predicted damage to local folding. In several of these regions, the model indicated loss of key structural features such as domain boundaries and loops that normally separate groups of genes and their switches. Strikingly, some of these hotspots were not especially heavily mutated overall; what set them apart was the severity of the folding disruption when variants did occur there. These regions contained genes involved in brain development and known cancer-related functions, suggesting that subtle 3D miswiring, rather than simple mutation counts, may be what matters most.
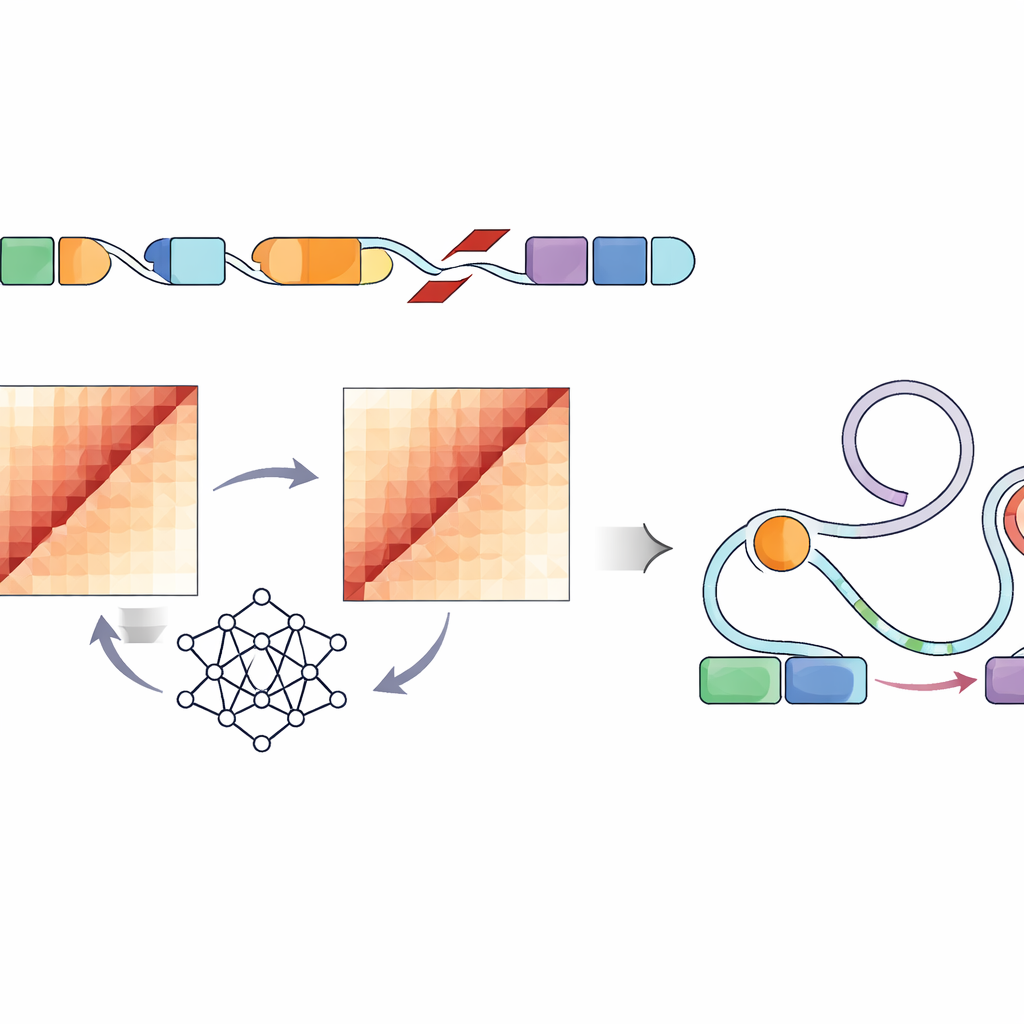
Linking Folding Disruptions to Gene Control Switches
Because not every change in DNA shape will affect how cells behave, the researchers next looked specifically at regulatory elements—enhancers marked by characteristic chemical tags and open chromatin in tumor-like cell lines. They found that highly disruptive variants in several pediatric tumor types were more likely to sit in or near these active enhancer regions. Building on an existing framework called the Activity-by-Contact model, they created an “ABC disruption score” that boosts variants predicted to disturb genome folding specifically where these enhancers reside. This refined scoring surfaced structural variants that strongly altered predicted contacts between enhancers and nearby genes with roles in cell growth, survival, and brain function, including well-known cancer genes such as PDGFRA, ID2, MYCN, and others.
Clues to New Cancer Drivers and Future Care
Focusing on a particularly aggressive tumor type called atypical teratoid/rhabdoid tumor, the method highlighted rearrangements near genes involved in chromatin remodeling, DNA repair, and neural development. In several cases, the tumors carrying these variants also showed unusually high or low expression of the nearby genes, consistent with enhancer “hijacking” through altered 3D contacts. While these findings still need experimental confirmation, they point to a powerful new way to sift through vast numbers of structural variants and prioritize those most likely to influence tumor behavior. In the long run, such machine-learning–guided maps of the genome’s folding landscape could help doctors interpret sequencing results from young cancer patients, uncover hidden drivers of disease, and ultimately guide the search for more precise, less toxic treatments.
Citation: Gjoni, K., Zhang, S., Yan, R.E. et al. Machine learning-predicted chromatin organization landscape across pediatric tumors. Sci Rep 16, 10790 (2026). https://doi.org/10.1038/s41598-026-44925-3
Keywords: pediatric brain tumors, structural variants, 3D genome organization, machine learning genomics, enhancer hijacking