Clear Sky Science · de
Durch maschinelles Lernen vorhergesagte Landschaft der Chromatinorganisation bei pädiatrischen Tumoren
Warum die Faltung des Genoms für kranke Kinder wichtig ist
Pädiatrische Hirntumoren gehören zu den tödlichsten Krebserkrankungen im Kindesalter, und bei vielen jungen Patientinnen und Patienten wissen die Ärzte noch immer nicht genau, was auf DNA‑Ebene schiefgelaufen ist. Diese Studie nimmt einen neuen Blickwinkel ein: nicht nur, welche Gene mutiert sind, sondern wie großflächige DNA‑Umordnungen die Art und Weise verändern, wie das Genom in jeder Zelle gefaltet ist. Durch die Kombination großer Krebsdatensätze mit einem leistungsfähigen Modell des maschinellen Lernens zeigen die Autorinnen und Autoren, dass versteckte Störungen in diesem 3D‑Faltungsmuster zur Entstehung kindlicher Tumoren beitragen können — und dass Computer nun die gefährlichsten Veränderungen für weitergehende Untersuchungen hervorheben können.
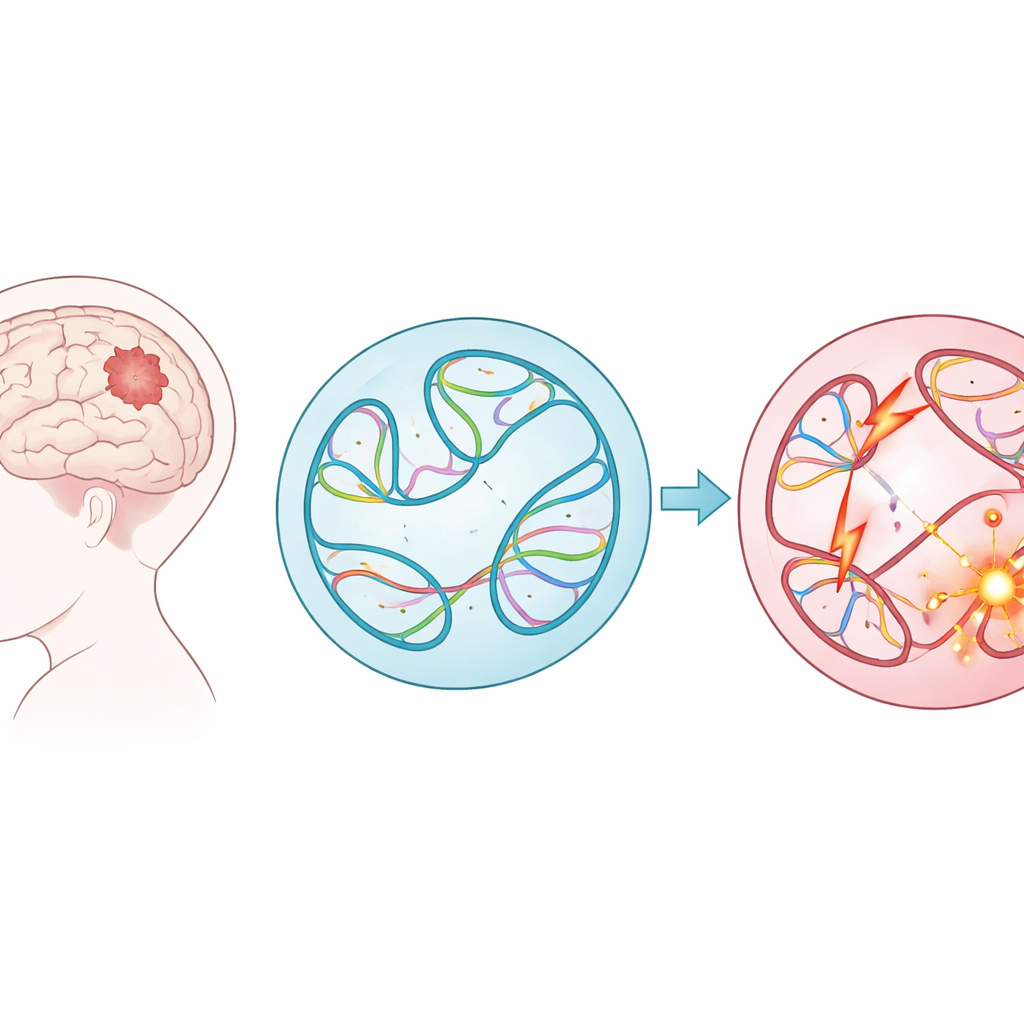
Große DNA‑Umbauten bei kindlichen Tumoren
Statt sich auf kleine Tippfehler im genetischen Code zu konzentrieren, untersuchten die Forschenden strukturelle Varianten — große Schnitte, Kopien, Umdrehungen und Einfügungen langer DNA‑Abschnitte. Diese großskaligen Veränderungen können Gene verschmelzen, schützende Regionen löschen oder starke Schaltstellen (Enhancer) neben die falschen Gene setzen. Mit Daten von fast 1.900 Kindern aus dem Children’s Brain Tumor Network, die 61 Tumortypen abdecken, katalogisierte das Team fast 300.000 solcher Varianten. Sie fanden, dass einige Tumorgruppen, etwa Lymphome und Sarkome, deutlich mehr dieser Veränderungen aufwiesen als andere, und dass Tumoren, die nach einer Erstbehandlung zurückkehrten oder fortschritten, typischerweise mehr strukturelle Varianten trugen als die ursprünglichen Tumoren.
Mit KI das Genom in 3D sehen
Direkt zu messen, wie jede strukturelle Variante die Faltung der DNA in einer Zelle verändert, würde mühsame Experimente an Hunderttausenden von Stellen erfordern — praktisch unmöglich in diesem Umfang. Stattdessen nutzten die Autorinnen und Autoren ein konvolutionales neuronales Netzwerk namens Akita, das über ihre SuPreMo‑Akita‑Pipeline zugänglich ist, um vorauszusagen, wie sich ein eine Million Basenpaare langer DNA‑Abschnitt in 3D faltet. Für jede strukturelle Variante simulierten sie die lokale DNA‑Sequenz mit und ohne die Veränderung, ließen das Modell Kontaktkarten vorhersagen — Muster, die zeigen, welche Teile des Genoms miteinander in Kontakt stehen — und verglichen diese Karten. Je größer der Unterschied, desto stärker wurde die Variante als Störung der Genomorganisation prognostiziert. So konnten sie Varianten über alle Tumoren hinweg danach einordnen, wie stark sie die normalen Faltungsmuster zu verbiegen oder zu zerstören erwarteten.
Hotspots, an denen die Faltung schiefgeht
Als das Team das Genom auf Stellen durchsuchte, die wiederholt von stark störenden Varianten getroffen wurden, entdeckten sie fünf rekurrent gestörte Regionen — DNA‑Abschnitte, in denen die Tumoren vieler Kinder verschiedener Typen starke vorhergesagte Schäden an der lokalen Faltung zeigten. In mehreren dieser Regionen zeigte das Modell den Verlust wichtiger struktureller Merkmale wie Domänengrenzen und Schleifen an, die normalerweise Gruppen von Genen und deren Schaltern trennen. Auffällig war, dass einige dieser Hotspots insgesamt nicht besonders stark mutiert waren; was sie auszeichnete, war die Schwere der Faltungsstörung, wenn Varianten dort auftraten. Diese Regionen enthielten Gene, die an der Gehirnentwicklung und an bekannten krebsrelevanten Funktionen beteiligt sind, was darauf hindeutet, dass subtile 3D‑Fehlverdrahtungen statt einfacher Mutationszahlen das Entscheidende sein könnten.
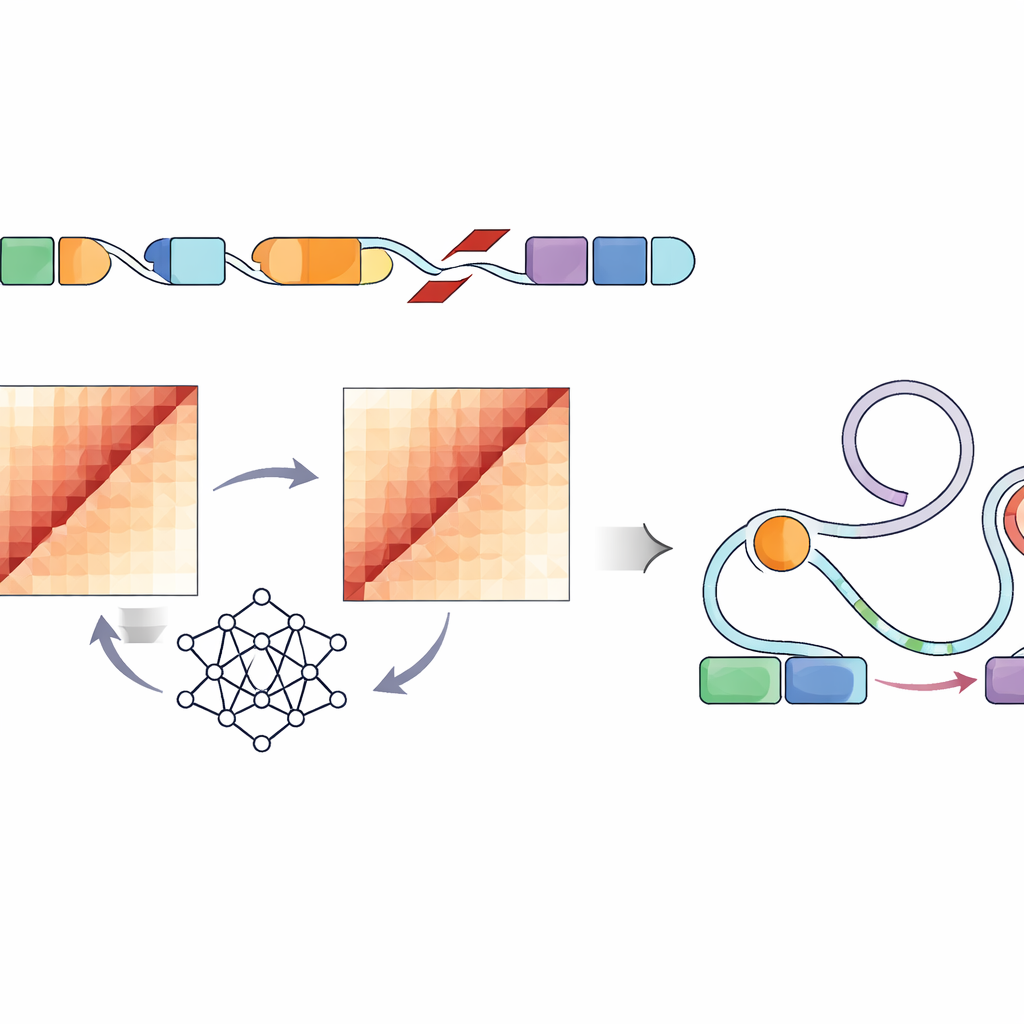
Verknüpfung von Faltungsstörungen mit Genregulations‑Schaltern
Da nicht jede Veränderung der DNA‑Form Auswirkungen auf das Verhalten von Zellen haben wird, betrachteten die Forschenden als Nächstes gezielt regulatorische Elemente — Enhancer, die durch charakteristische chemische Markierungen und offene Chromatinbereiche in tumornahen Zelllinien gekennzeichnet sind. Sie fanden, dass stark störende Varianten in mehreren pädiatrischen Tumortypen wahrscheinlicher in oder in der Nähe dieser aktiven Enhancer‑Regionen lagen. Aufbauend auf einem bestehenden Rahmenwerk, dem Activity‑by‑Contact‑Modell, entwickelten sie einen „ABC‑Disruption‑Score“, der Varianten stärker gewichtet, die die Genomfaltung speziell dort stören, wo diese Enhancer sitzen. Diese verfeinerte Bewertung hob strukturelle Varianten hervor, die vorhergesagte Kontakte zwischen Enhancern und nahegelegenen Genen stark veränderten — Gene, die an Zellwachstum, Überleben und Gehirnfunktionen beteiligt sind, darunter bekannte Krebsgene wie PDGFRA, ID2, MYCN und andere.
Hinweise auf neue Krebs‑Treiber und künftige Versorgung
Mit Blick auf einen besonders aggressiven Tumortyp, das atypische teratoid/rhabdoide Tumor, machte die Methode Rearrangements neben Genen sichtbar, die an Chromatin‑Remodeling, DNA‑Reparatur und neuronaler Entwicklung beteiligt sind. In mehreren Fällen zeigten die Tumoren mit diesen Varianten auch ungewöhnlich hohe oder niedrige Expression der benachbarten Gene, was mit einem Enhancer‑„Hijacking“ durch veränderte 3D‑Kontakte vereinbar ist. Während diese Befunde noch experimentell bestätigt werden müssen, deuten sie auf einen kraftvollen neuen Weg hin, riesige Mengen struktureller Varianten zu durchsieben und diejenigen zu priorisieren, die am ehesten das Tumorverhalten beeinflussen. Langfristig könnten solche durch maschinelles Lernen geführten Karten der Faltungslandschaft des Genoms Ärzten helfen, Sequenzierungsergebnisse junger Krebspatientinnen und -patienten besser zu interpretieren, verborgene Treiber der Krankheit aufzudecken und letztlich die Suche nach präziseren, weniger toxischen Therapien zu lenken.
Zitation: Gjoni, K., Zhang, S., Yan, R.E. et al. Machine learning-predicted chromatin organization landscape across pediatric tumors. Sci Rep 16, 10790 (2026). https://doi.org/10.1038/s41598-026-44925-3
Schlüsselwörter: pädiatrische Hirntumoren, strukturelle Varianten, 3D-Genomorganisation, maschinelles Lernen Genomik, Enhancer-Hijacking