Clear Sky Science · fr
Organisation de la chromatine prédite par apprentissage automatique dans les tumeurs pédiatriques
Pourquoi le repliement du génome compte pour les enfants malades
Les tumeurs cérébrales pédiatriques font partie des cancers infantiles les plus meurtriers, et pourtant, pour de nombreux jeunes patients, les médecins ne savent toujours pas précisément ce qui a mal tourné dans leur ADN. Cette étude explore un angle nouveau : pas seulement quelles gènes sont mutés, mais comment de vastes réarrangements d’ADN modifient la façon dont le génome se replie à l’intérieur de chaque cellule. En combinant de larges jeux de données sur le cancer avec un modèle d’apprentissage automatique puissant, les auteurs montrent que des perturbations cachées de ce pliage 3D peuvent contribuer aux tumeurs infantiles — et que les ordinateurs peuvent désormais signaler les changements les plus dangereux pour des études complémentaires.
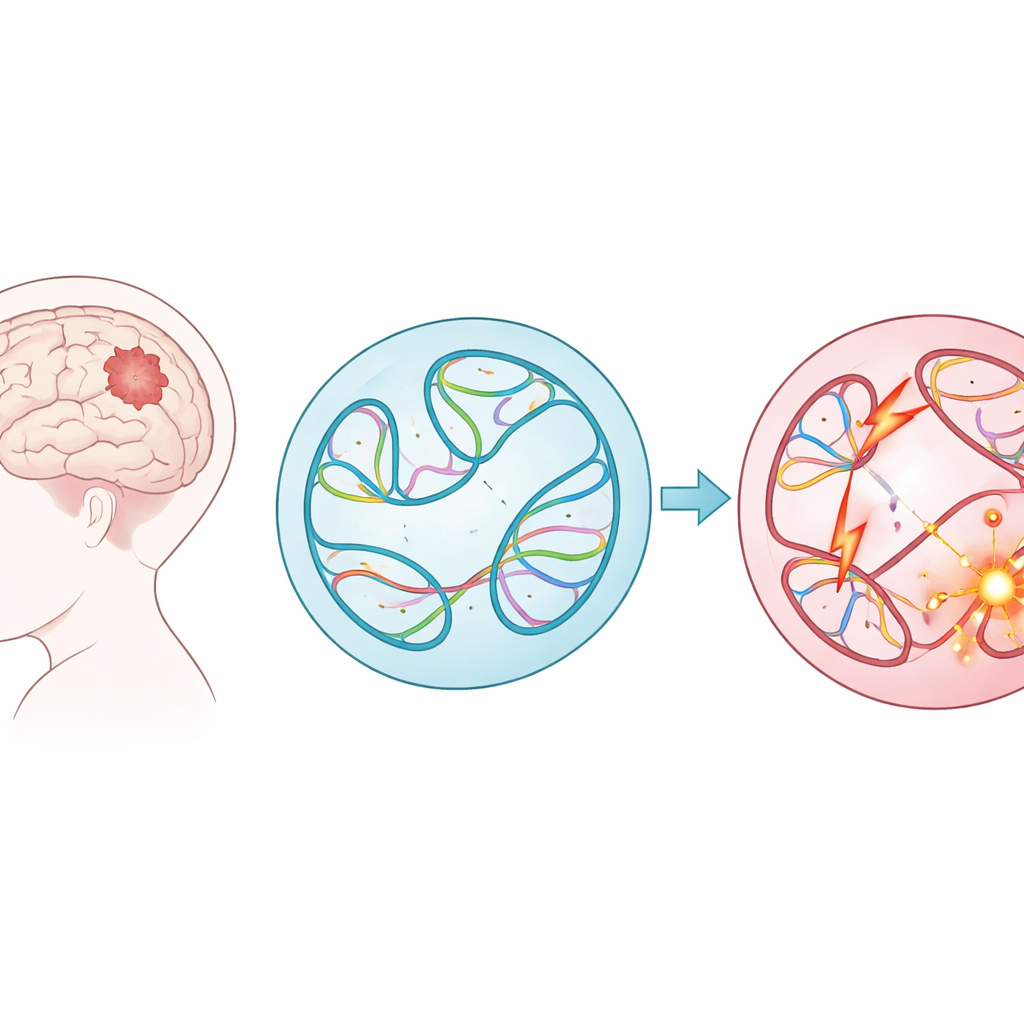
Vastes remaniements de l’ADN dans les tumeurs de l’enfance
Plutôt que de se concentrer sur de petites fautes dans le code génétique, les chercheurs ont étudié les variants structurels — de grandes coupures, duplications, inversions et collages de longs segments d’ADN. Ces changements à grande échelle peuvent fusionner des gènes, supprimer des régions protectrices ou placer des interrupteurs puissants (enhancers) à côté des mauvais gènes. En utilisant des données provenant de près de 1 900 enfants du Children’s Brain Tumor Network, couvrant 61 types de tumeurs, l’équipe a répertorié presque 300 000 de ces variants. Ils ont constaté que certains groupes tumoraux, comme les lymphomes et les sarcomes, présentaient beaucoup plus de ces altérations que d’autres, et que les tumeurs récidivantes ou progressives après le traitement initial comportaient généralement davantage de variants structurels que les tumeurs d’origine.
Utiliser l’IA pour voir le génome en 3D
Mesurer directement comment chaque variant structurel modifie le repliement de l’ADN dans une cellule nécessiterait des expériences laborieuses pour des centaines de milliers de sites — pratiquement impossible à cette échelle. À la place, les auteurs ont utilisé un réseau de neurones convolutif appelé Akita, via leur pipeline SuPreMo-Akita, pour prédire comment un segment d’un million de paires de bases d’ADN se replie en 3D. Pour chaque variant structurel, ils ont simulé la séquence d’ADN locale avec et sans la modification, demandé au modèle de prédire des cartes de contacts — des schémas montrant quelles parties du génome se touchent — puis comparé ces cartes. Plus la différence était grande, plus le variant était prévu pour perturber l’organisation du génome. Cela leur a permis de classer les variants à travers toutes les tumeurs selon l’intensité de leur impact attendu sur les motifs de repliement normaux.
Points chauds où le repliement déraille
En parcourant le génome à la recherche d’endroits touchés à plusieurs reprises par des variants fortement perturbateurs, l’équipe a mis au jour cinq régions récurrentement altérées — des segments d’ADN où de nombreuses tumeurs d’enfants, de types variés, montraient de fortes perturbations prédictives du repliement local. Dans plusieurs de ces régions, le modèle indiquait la perte de caractéristiques structurelles clés telles que des frontières de domaines et des boucles qui séparent normalement des groupes de gènes et leurs régulateurs. Fait frappant, certains de ces points chauds n’étaient pas particulièrement fortement mutés dans l’absolu ; ce qui les distinguait était la sévérité de la perturbation du repliement lorsqu’un variant y survenait. Ces régions contenaient des gènes impliqués dans le développement cérébral et des fonctions liées au cancer, ce qui suggère qu’un mauvais câblage 3D subtil, plutôt que le simple nombre de mutations, peut être ce qui importe le plus.
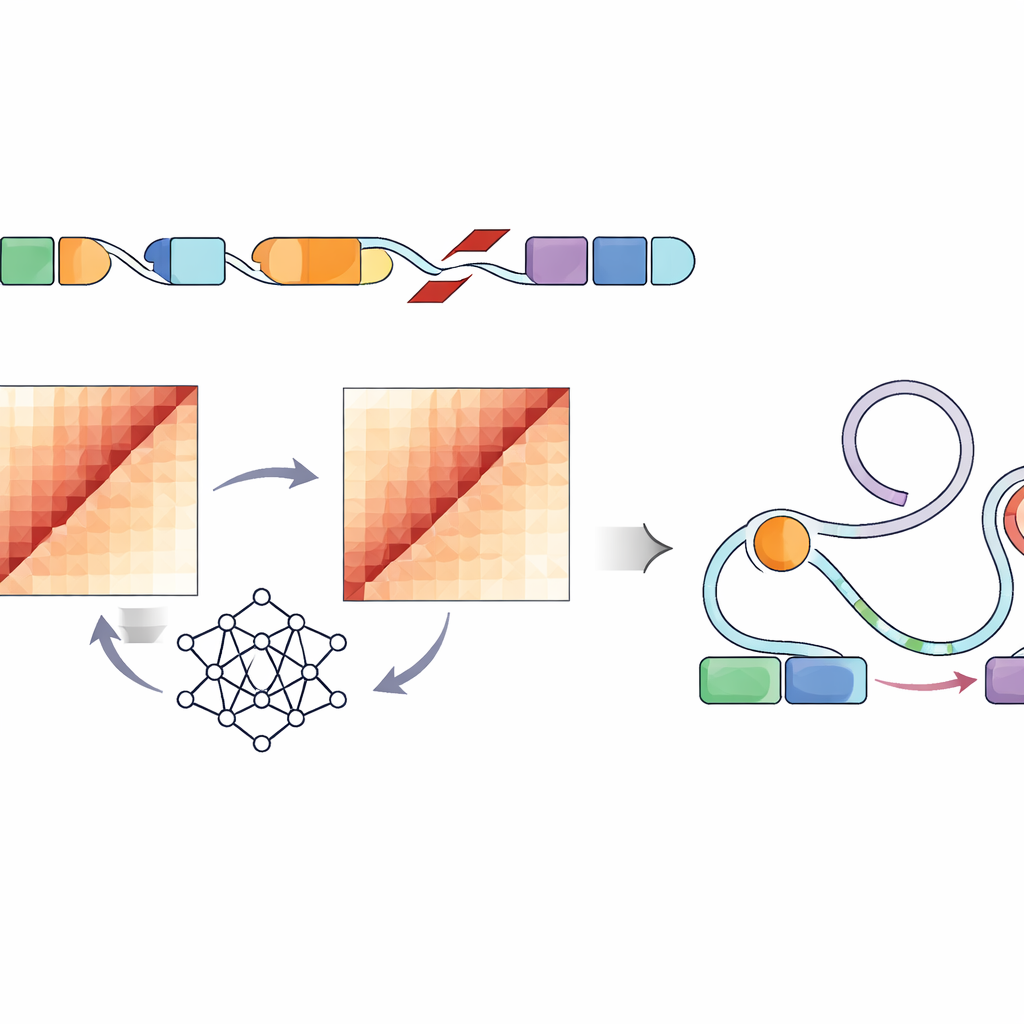
Relier les perturbations du repliement aux interrupteurs de contrôle des gènes
Parce que tous les changements de forme de l’ADN n’affecteront pas le comportement cellulaire, les chercheurs se sont ensuite focalisés sur les éléments régulateurs — des enhancers marqués par des balises chimiques caractéristiques et une chromatine ouverte dans des lignées cellulaires de type tumoral. Ils ont trouvé que les variants fortement perturbateurs dans plusieurs types de tumeurs pédiatriques avaient plus de chances de se situer dans ou à proximité de ces régions d’enhancers actives. En s’appuyant sur un cadre existant appelé modèle Activity-by-Contact (ABC), ils ont créé un « score de perturbation ABC » qui met en avant les variants prédits pour perturber le repliement du génome précisément là où ces enhancers résident. Cette pondération affinée a mis en lumière des variants structurels modifiant fortement les contacts prédits entre enhancers et gènes voisins impliqués dans la croissance cellulaire, la survie et la fonction cérébrale, y compris des gènes bien connus du cancer tels que PDGFRA, ID2, MYCN et d’autres.
Indices de nouveaux drivers du cancer et perspectives pour les soins
En se concentrant sur un type tumoral particulièrement agressif appelé tumeur teratoïde/rhabdoïde atypique, la méthode a mis en évidence des réarrangements proches de gènes impliqués dans le remodelage de la chromatine, la réparation de l’ADN et le développement neural. Dans plusieurs cas, les tumeurs portant ces variants montraient également une expression anormalement élevée ou faible des gènes voisins, cohérente avec un « détournement » d’enhancer via des contacts 3D modifiés. Bien que ces résultats nécessitent encore une confirmation expérimentale, ils indiquent une nouvelle manière puissante de trier d’énormes quantités de variants structurels et de prioriser ceux qui sont le plus susceptibles d’influencer le comportement tumoral. À long terme, de telles cartes guidées par l’apprentissage automatique du paysage de repliement du génome pourraient aider les médecins à interpréter les résultats de séquençage chez les jeunes patients atteints de cancer, à découvrir des drivers cachés de la maladie et, finalement, à orienter la recherche vers des traitements plus précis et moins toxiques.
Citation: Gjoni, K., Zhang, S., Yan, R.E. et al. Machine learning-predicted chromatin organization landscape across pediatric tumors. Sci Rep 16, 10790 (2026). https://doi.org/10.1038/s41598-026-44925-3
Mots-clés: tumeurs cérébrales pédiatriques, variants structurels, organisation 3D du génome, génomique et apprentissage automatique, détournement d’enhancers