Clear Sky Science · pl
Wirusowy antagonista APOBEC3 rozróżnia HHV-6A od HHV-6B
Dlaczego niektóre powszechne wirusy wywołują choroby, a inne pozostają ciche
Większość dorosłych nosi przez całe życie kilka herpeswirusów, lecz tylko niektóre z tych pasażerów wywołują poważne choroby. To badanie stawia proste pytanie o dalekosiężnych konsekwencjach: dlaczego jeden przedstawiciel rodziny ludzkiego herpeswirusa 6 (HHV-6B) często powoduje choroby u niemowląt i pacjentów po przeszczepach, podczas gdy jego niemal bliźniaczy odpowiednik (HHV-6A) zwykle pozostaje niezauważony? Śledząc, jak te wirusy zderzają się z wbudowanym w komórkę systemem obrony DNA, autorzy odkrywają molekularną przepychankę, która pomaga wyjaśnić ich diametralnie różne zachowanie kliniczne. 
Opowieść o dwóch blisko spokrewnionych wirusach
HHV-6A i HHV-6B to bardzo podobne wirusy, które atakują ten sam typ komórek odpornościowych i dzielą ponad 90% genów. Mimo to zachowują się inaczej u ludzi. HHV-6B jest główną przyczyną rumienia nagłego (roseoli), powszechnej dziecięcej wysypki, która niekiedy prowadzi do zapalenia mózgu, zwłaszcza u pacjentów poddawanych terapiom szpiku kostnego lub zaawansowanym terapiom komórkami T. HHV-6A natomiast zwykle pozostaje cichy i rzadko wiąże się z jednoznacznymi chorobami. Ponieważ oba wirusy używają znanych receptorów do wejścia do komórek, autorzy podejrzewali, że to, co dzieje się po wejściu — w obrębie mechanizmów obsługi DNA w komórce — może być kluczem do ich różnych skutków.
Wbudowany w komórkę edytor DNA wkracza do akcji
Nasze komórki wytwarzają grupę enzymów zwanych białkami APOBEC3, które patrolują DNA i mogą sabotować genom wirusowy poprzez chemiczną konwersję niektórych zasad DNA, wywołując burzę mutacji. Porównując aktywność genów w różnych liniach komórek T, zespół zauważył, że kilka genów APOBEC3 jest bardziej aktywnych w komórkach, które dopuszczają wzrost HHV-6B, ale nie HHV-6A. Gdy zsekwencjonowali bogaty w GC fragment wirusowego DNA z zakażonych komórek, HHV-6A nosiło ciężki ładunek mutacji typowych dla APOBEC, podczas gdy HHV-6B pozostało w dużej mierze nienaruszone. Wyłączenie poszczególnych genów APOBEC3, zwłaszcza APOBEC3B i APOBEC3C, zwiększało replikację HHV-6A i ostro zmniejszało jego obciążenie mutacyjne, co pokazuje, że ten system obronny gospodarza rzeczywiście ogranicza HHV-6A. HHV-6B w tych samych komórkach w dużym stopniu uchodził spod tej edycji DNA.
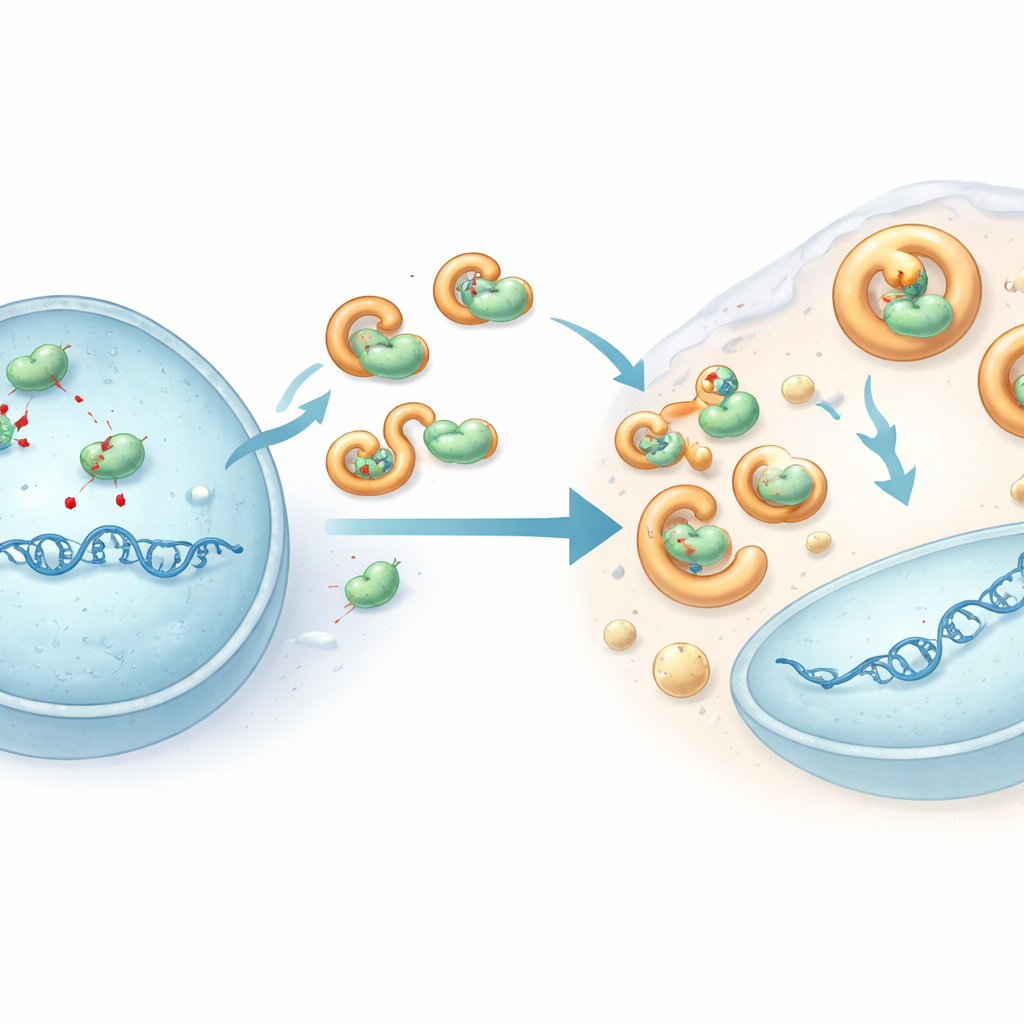
Wirusowa tarcza, która unieszkodliwia edytora
Aby zrozumieć, jak HHV-6B uchodzi przed tym atakiem, badacze skupili się na jednym białku wirusowym zwanym U28, zmodyfikowanej wersji enzymu używanego przez wiele herpeswirusów do budowy DNA. W komórkach zakażonych HHV-6B APOBEC3B i APOBEC3C były wypychane z jądra komórkowego (gdzie kopiowane jest DNA wirusa) do płynu otaczającego i ich całkowite poziomy spadały. Doświadczenia w komórkach inżynieryjnych wykazały, że białko U28 HHV-6B fizycznie wiąże się z kilkoma białkami APOBEC3, przyciąga je do przypominających ciecz kropli powiązanych ze szlakami usuwania komórkowego i sprzyja ich rozkładowi. To sekwestracja trzyma enzymy z dala od DNA wirusa i zapobiega ich wyrządzaniu rozległych szkód. Gdy U28 zostało obniżone w zakażonych komórkach, poziomy APOBEC3 odbijały się i genom HHV-6B gromadził więcej mutacji, co potwierdza, że U28 działa jak zbroja przeciw tej antywirusowej edycji.
Niewielkie różnice w ekspresji, wielkie różnice w rezultacie
Ciekawie, białko U28 z HHV-6A jest niemal identyczne sekwencyjnie z wersją z HHV-6B i może również wiązać oraz obniżać poziomy APOBEC3, gdy jest sztucznie nadprodukowane. Kluczowa różnica dotyczy ilości, nie jakości: podczas naturalnego zakażenia HHV-6B uruchamia U28 znacznie silniej niż HHV-6A. Pomiary w zakażeniach laboratoryjnych i w pierwotnych komórkach krwi wykazały znacznie wyższe poziomy RNA U28 w HHV-6B, wraz z lepszym zachowaniem jego genomu. U rzadkiego pacjenta z reaktywacją wrodzonego genomu HHV-6A zespół wykrył uderzająco zróżnicowaną pulę wariantów HHV-6A wypełnionych mutacjami przypominającymi APOBEC, w przeciwieństwie do relatywnie stabilnych sekwencji HHV-6B obserwowanych u pacjentów po przeszczepach i z zapaleniem mózgu. Autorzy proponują, że słaba ekspresja U28 u HHV-6A pozostawia go wrażliwym na edytor DNA komórki, ograniczając jego zdolność do efektywnej replikacji i rozprzestrzeniania się.
Co to oznacza dla zdrowia i chorób
Z perspektywy ogólnej, HHV-6B nauczył się unieszkodliwiać kluczowy alarm komórkowy, co pozwala mu kopiować DNA bez uszkodzeń i ustanawiać silniejsze, niekiedy szkodliwe zakażenia. HHV-6A, które nie potrafi w pełni uciszyć tej obrony, zostaje mocno naznaczone mutacjami i w rezultacie może mniej efektywnie się replikować oraz powodować mniej widocznych chorób. Łącząc te różne wyniki z jednym wirusowym przełącznikiem kontrolującym siłę produkcji U28, badanie oferuje konkretną molekularną wyjaśnienie, dlaczego dwa niemal identyczne wirusy mogą mieć tak różne ślady kliniczne. Podkreśla też enzymy APOBEC3 jako potencjalnych kształtujących ewolucję wirusów i jako możliwe dźwignie dla przyszłych strategii antywirusowych, które albo wzmocnią edytor komórkowy przeciw niebezpiecznym wirusom, albo pomogą zrozumieć, dlaczego niektóre infekcje pozostają łagodne.
Cytowanie: Arii, J., Aktar, S., Huang, J.R. et al. A viral APOBEC3 antagonist distinguishes HHV-6A from HHV-6B. Nat Commun 17, 3566 (2026). https://doi.org/10.1038/s41467-026-71951-6
Słowa kluczowe: ludzki herpeswirus 6, APOBEC3, unikanie układu odpornościowego przez wirusy, mutacje wirusowe, białko U28