Clear Sky Science · en
A viral APOBEC3 antagonist distinguishes HHV-6A from HHV-6B
Why some common viruses make us sick and others stay quiet
Most adults carry several herpesviruses for life, but only some of these stowaways ever cause serious disease. This study asks a simple question with far-reaching implications: why does one member of the human herpesvirus 6 family (HHV-6B) often make infants and transplant patients ill, while its near-twin (HHV-6A) usually lurks unnoticed? By tracing how these viruses clash with a built‑in DNA defense system inside our cells, the authors uncover a molecular tug‑of‑war that helps explain their very different clinical behavior. 
A tale of two closely related viruses
HHV-6A and HHV-6B are extremely similar viruses that target the same kinds of immune cells and share more than 90% of their genes. Yet they behave differently in people. HHV-6B is the main cause of roseola, a common childhood rash that can sometimes lead to brain inflammation, especially in patients undergoing bone‑marrow or advanced T‑cell therapies. HHV-6A, in contrast, is usually silent and rarely linked to clear disease. Because both viruses use known receptors to enter cells, the authors suspected that what happens after entry—inside the cell’s own DNA-handling machinery—might be the key to their different impact.
The cell’s built‑in DNA editor steps in
Our cells express a group of enzymes called APOBEC3 proteins that patrol DNA and can sabotage viral genomes by chemically converting certain DNA letters, triggering a storm of mutations. By comparing gene activity in different T‑cell lines, the team noticed that several APOBEC3 genes were more active in cells that allow HHV-6B, but not HHV-6A, to grow. When they sequenced a GC‑rich stretch of viral DNA from infected cells, HHV-6A carried a heavy burden of APOBEC‑style mutations, while HHV-6B remained mostly intact. Turning off individual APOBEC3 genes, especially APOBEC3B and APOBEC3C, boosted HHV-6A replication and sharply reduced its mutation load, showing that this host defense system genuinely restricts HHV-6A. HHV-6B, however, largely escaped this DNA editing in the same cells.
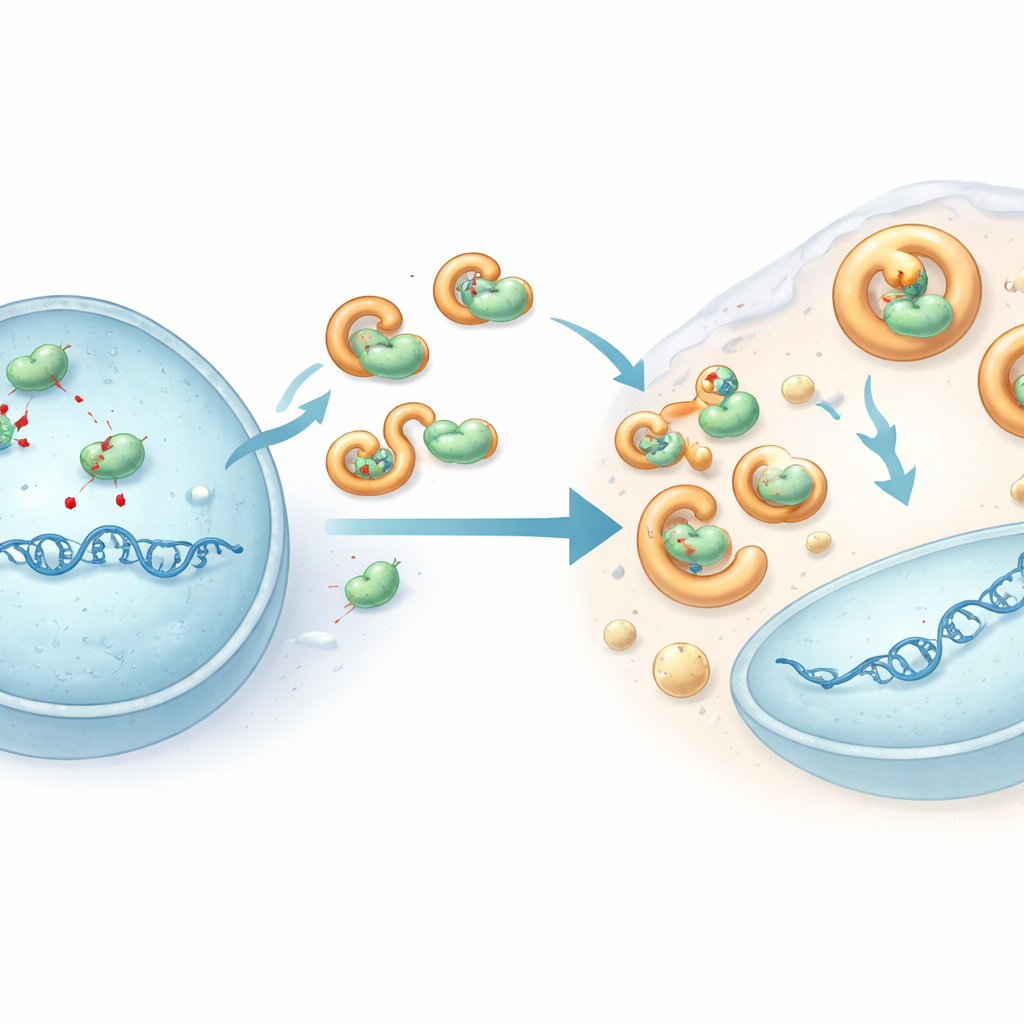
A viral shield that disarms the editor
To understand how HHV-6B dodges this attack, the researchers focused on one viral protein called U28, a modified version of an enzyme that many herpesviruses use for DNA building. In HHV-6B‑infected cells, APOBEC3B and APOBEC3C were pushed out of the nucleus (where viral DNA is copied) into the surrounding fluid and their overall levels dropped. Experiments in engineered cells showed that the HHV-6B U28 protein physically latches onto several APOBEC3 proteins, pulls them into liquid‑like droplets connected to cellular disposal pathways, and promotes their breakdown. This sequestration keeps the enzymes away from viral DNA and prevents them from inflicting widespread damage. When U28 was reduced in infected cells, APOBEC3 levels bounced back and HHV-6B genomes accumulated more mutations, confirming that U28 acts as an armor against this antiviral editing.
Small differences in expression, big differences in outcome
Curiously, the U28 protein from HHV-6A is almost identical in sequence to that from HHV-6B and can also bind and lower APOBEC3 proteins when artificially overproduced. The crucial distinction is quantity, not quality: during natural infection, HHV-6B turns on U28 much more strongly than HHV-6A. Measurements in laboratory infections and in primary blood cells showed far higher U28 RNA levels in HHV-6B, along with better preservation of its genome. In a rare patient with reactivation of an inherited HHV-6A genome, the team detected a strikingly diverse swarm of HHV-6A variants packed with APOBEC‑like mutations, in contrast to the relatively stable HHV-6B sequences seen in transplant and encephalitis patients. The authors propose that HHV-6A’s weak U28 expression leaves it vulnerable to the cell’s DNA editor, limiting its ability to replicate efficiently and spread.
What this means for health and disease
Seen from a lay perspective, HHV-6B has learned to disarm a key cellular alarm, allowing it to copy its DNA cleanly and establish stronger, sometimes harmful infections. HHV-6A, which fails to fully silence this defense, becomes heavily scarred by mutations and may therefore replicate less and cause fewer overt illnesses. By tying these outcomes to a single viral switch that controls how strongly U28 is produced, the study offers a concrete molecular explanation for why two near‑identical viruses can have such different clinical footprints. It also highlights APOBEC3 enzymes as potential shapers of viral evolution and as possible levers for future antiviral strategies that aim either to reinforce the cell’s editor against dangerous viruses or to understand why some infections remain mild.
Citation: Arii, J., Aktar, S., Huang, J.R. et al. A viral APOBEC3 antagonist distinguishes HHV-6A from HHV-6B. Nat Commun 17, 3566 (2026). https://doi.org/10.1038/s41467-026-71951-6
Keywords: human herpesvirus 6, APOBEC3, viral immune evasion, virus mutation, U28 protein