Clear Sky Science · pl
Cytozolowa funkcja dodatkowa ACAD9: tłumienie osteoklastogenezy zależnej od TRAF6 i ochrona przed osteoporozą
Dlaczego kości słabną z wiekiem
Złamania szyjki kości udowej i kruchy kręgosłup należą do najbardziej obawianych problemów związanych ze starzeniem, lecz ukryta biologia komórkowa stojąca za tymi złamaniami rzadko pojawia się poza laboratorium. To badanie ujawnia zaskakującą rolę białka mitochondrialnego nazwanego ACAD9 w utrzymaniu wytrzymałości kości. Działając zarówno w „elektrowniach” komórkowych, jak i w układzie sygnałowym komórki, ACAD9 pomaga powstrzymywać komórki „jedzące kość” i może otworzyć nową drogę zapobiegania osteoporozie.
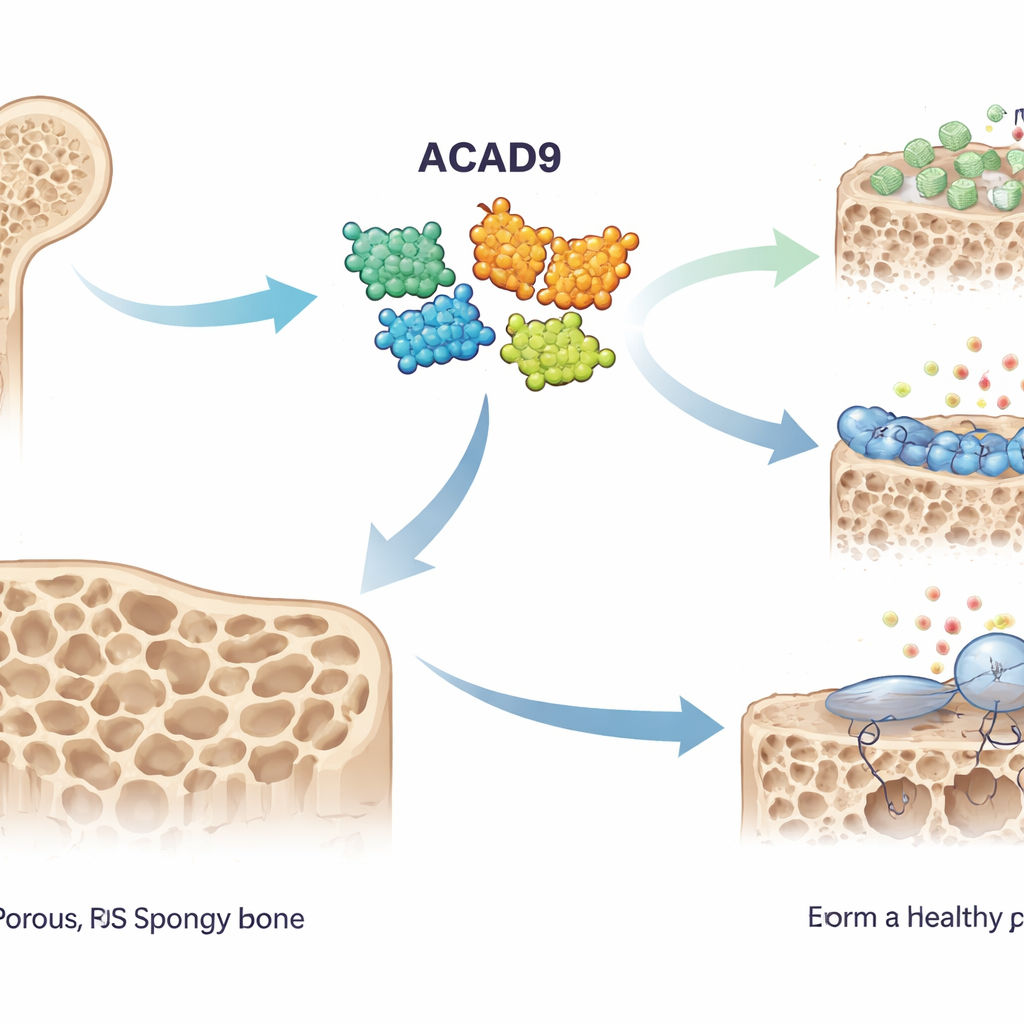
Budowniczowie i pożeracze kości
Zdrowa kość jest nieustannie przebudowywana przez dwie przeciwstawne grupy komórek: osteoblasty, które budują kość, i osteoklasty, które ją rozpuszczają. W osteoporozie równowaga przesuwa się na korzyść „pożeraczy”, pozostawiając szkielet cienkim i kruchym. Osteoklasty aktywowane są, gdy sygnał zwany RANKL łączy się ze swoim receptorem na komórkach prekursorowych, uruchamiając białko o nazwie TRAF6. To z kolei wyzwala kaskady chemiczne i wyrzut reaktywnych form tlenu — wysoko reaktywnych molekuł często określanych jako ROS — które skłaniają prekursorowe komórki do fuzji w duże, resorbujące kość komórki. Autorzy przypuszczali, że ACAD9, znany z roli w produkcji energii w mitochondriach, może także wpływać na ten proces.
Mitochondrialny strażnik przed stresem
W mitochondriach ACAD9 pomaga montować duży kompleks białkowy zwany kompleksem I i sprzyja tworzeniu „superkompleksów”, uporządkowanych zespołów kilku kompleksów łańcucha oddechowego. Struktury te zwiększają wydajność produkcji energii i ograniczają niepożądane ROS. W eksperymentach komórkowych badacze zaobserwowali, że gdy prekursory osteoklastów były wystawione na RANKL i zaczynały dojrzewać, poziomy ACAD9 spadały, kompleks I i superkompleksy się zmniejszały, a ROS gwałtownie rosły. Zmniejszenie ekspresji ACAD9 jeszcze bardziej wzmagało ROS i przyspieszało powstawanie osteoklastów, podczas gdy zwiększenie poziomu ACAD9 przywracało kompleks I, podnosiło energię komórkową (ATP), redukowało ROS i hamowało różnicowanie osteoklastów. Usuwanie ROS za pomocą leku przeciwutleniającego tylko częściowo odwracało skutki utraty ACAD9, co sugeruje, że ACAD9 działa także poprzez inną, nie‑mitochondrialną drogę.
ACAD9 jako molekularny hamulec dla pożeraczy kości
Zespół przyjrzał się następnie TRAF6, kluczowemu przekaźnikowi, którego RANKL używa do włączania genów osteoklastów. Odkryli, że część białka ACAD9 znajduje się w cytosolu zamiast w mitochondriach, gdzie może fizycznie wiązać się z TRAF6. To wiązanie zmienia sposób, w jaki TRAF6 współdziała ze swoimi enzymami partnerami, które przyłączają małe znaczniki „ubikwityny”. Bez ACAD9 TRAF6 staje się silnie ozdobiony jednym typem łańcucha ubikwityny (wiązanym przez pozycję zwaną K63), który działa jak rusztowanie aktywujące sygnały dalej w dół, w tym szlaki MAPK i NF‑κB napędzające wzrost osteoklastów i resorpcję kości. Obecność ACAD9 ogranicza te łańcuchy K63, natomiast sprzyja innemu typowi łańcuchów ubikwityny (wiązanym przez K48), kierując TRAF6 do zniszczenia przez komórkowe „niszczarki” białek. W istocie ACAD9 osłabia zdolność TRAF6 do sygnalizowania i przyspiesza jego usuwanie.
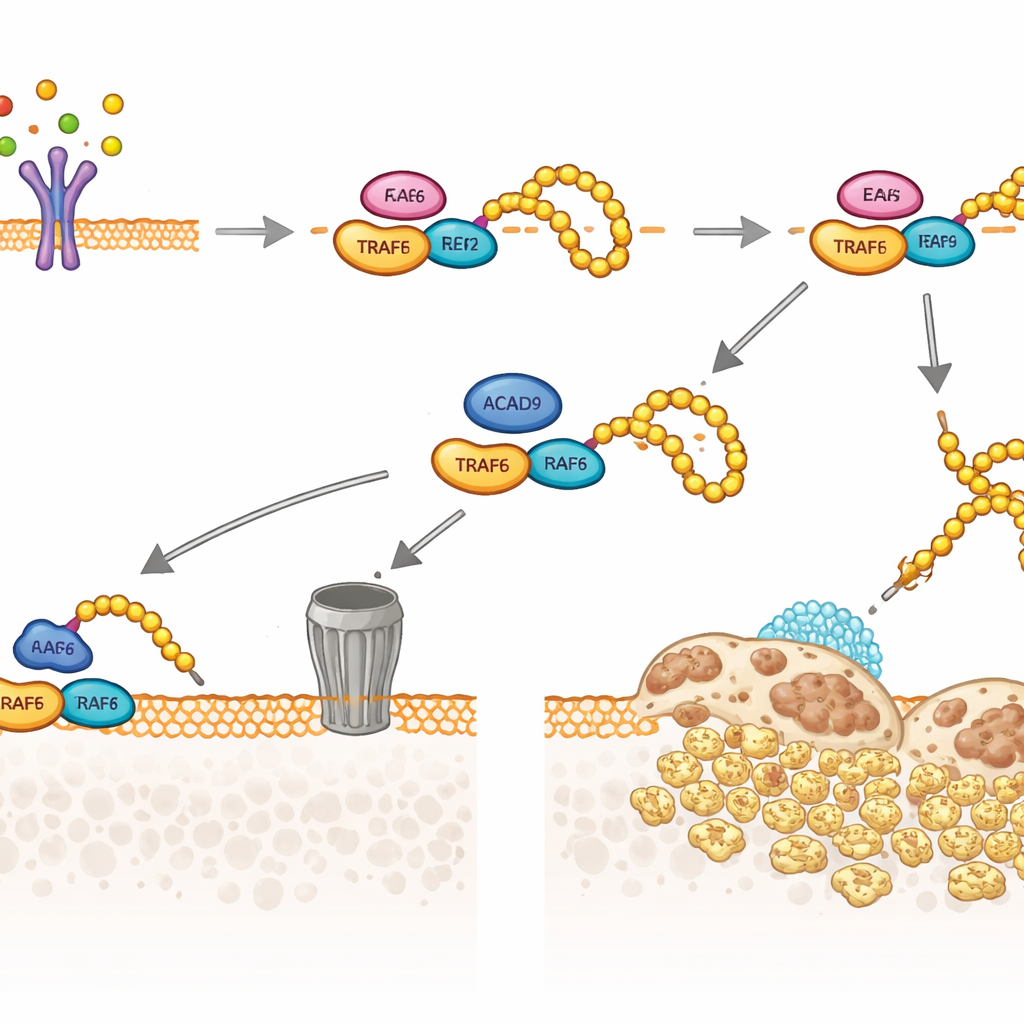
Od kości myszy do złamań u ludzi
Aby sprawdzić, jak to działa w żywym organizmie, badacze zaprojektowali myszy pozbawione ACAD9 specyficznie w prekursorach osteoklastów. W miarę starzenia się tych zwierząt tomografie ukazywały stopniowe przerzedzanie kości beleczkowej, większe odstępy między beleczkami i gorszą architekturę kości — wszystkie cechy charakterystyczne dla osteoporozy. Ich kości zawierały więcej osteoklastów i wyższe poziomy markerów resorpcji kości, podczas gdy kompleks I mitochondrialny, superkompleksy i ATP były zmniejszone. Cząsteczki sygnałowe pochodne TRAF6 były nadaktywne, a enzymy produkujące ROS podwyższone. Co ważne, aktywność tworzenia kości pozostała w dużej mierze normalna, co wskazuje, że nadmierna utrata kości wynikała głównie z nadaktywności osteoklastów. Dopełniając badania na myszach, analiza dużych danych z biobanków ludzkich powiązała rzadkie, szkodliwe warianty w genie ACAD9 z wyższym ryzykiem złamań kręgosłupa, sugerując znaczenie kliniczne.
Co to znaczy dla przyszłych terapii
Podsumowując, wyniki przedstawiają ACAD9 jako podwójnego obrońcę przed osteoporozą. W mitochondriach utrzymuje wydajną produkcję energii i kontroluje stres oksydacyjny; w cytosolu tłumi TRAF6, główny wyłącznik aktywacji osteoklastów, poprzez przekształcanie jego znakowania ubikwitynowego i promowanie jego degradacji. Gdy ACAD9 jest nieobecny lub obniżony, komórki resorbujące kość mnożą się, a kości słabną. Leki zwiększające aktywność ACAD9 lub naśladujące jego interakcję z TRAF6 mogłyby więc zaoferować nową, dwukierunkową strategię ochrony starzejących się kości — jednocześnie poprawiając metabolizm komórek i tłumiąc nadmierną resorpcję kości.
Cytowanie: Wang, M., Yuan, C., Zhang, Y. et al. Moonlighting cytosolic function of ACAD9: suppression of TRAF6-mediated osteoclastogenesis and protection against osteoporosis. Cell Death Dis 17, 362 (2026). https://doi.org/10.1038/s41419-026-08626-z
Słowa kluczowe: osteoporoza, osteoklasty, mitochondria, ubikwitynacja, remodelowanie kości