Clear Sky Science · de
Moonlighting-zytosolische Funktion von ACAD9: Unterdrückung der TRAF6-vermittelten Osteoklastogenese und Schutz vor Osteoporose
Warum Knochen mit dem Alter schwächer werden
Hüftbrüche und brüchige Wirbel gehören zu den gefürchtetsten Problemen des Alterns, doch die zugrunde liegende Zellbiologie hinter diesen Frakturen wird außerhalb des Labors selten besprochen. Diese Studie enthüllt eine überraschende Rolle eines mitochondrialen Proteins namens ACAD9 bei der Erhaltung unserer Knochengesundheit. Indem es sowohl in den Energiezentralen der Zelle als auch in deren Signalübertragung wirkt, hilft ACAD9, die knochenabbauenden Zellen in Schach zu halten, und könnte einen neuen Ansatz zur Prävention von Osteoporose eröffnen.
Knochenbauer und Knochenfresser
Gesunder Knochen wird fortlaufend von zwei gegensätzlichen Zellgruppen umgebaut: Osteoblasten, die Knochen aufbauen, und Osteoklasten, die ihn auflösen. Bei Osteoporose verschiebt sich das Gleichgewicht zugunsten der Knochenfresser, sodass das Skelett dünn und brüchig wird. Osteoklasten werden aktiviert, wenn ein Signal namens RANKL an seinen Rezeptor auf Vorläuferzellen bindet und das Protein TRAF6 einschaltet. Das löst chemische Kaskaden und eine Welle reaktiver Sauerstoffspezies (ROS) aus — hochreaktive Moleküle, die diese Vorläufer zur Fusion zu großen, knochenresorbierenden Zellen treiben. Die Autoren vermuteten, dass ACAD9, bekannt für seine Rolle bei der mitochondrialen Energieproduktion, diesen Prozess ebenfalls beeinflussen könnte.
Ein mitochondrialer Wächter gegen Stress
In den Mitochondrien hilft ACAD9 beim Zusammenbau einer großen Proteinanlage, des Komplex I, und fördert die Bildung von „Superkomplexen“, geordneten Zusammenschlüssen mehrerer Atmungskettenkomplexe. Diese Strukturen machen die Energieproduktion effizienter und begrenzen unerwünschte ROS. In Zellversuchen fanden die Forschenden heraus, dass bei Exposition von Osteoklastvorläufern gegenüber RANKL und während ihrer Reifung die ACAD9-Spiegel sanken, Komplex I und Superkomplexe abnahmen und ROS stark anstiegen. Das Herunterregulieren von ACAD9 verstärkte ROS weiter und beschleunigte die Osteoklastenbildung, während eine Erhöhung von ACAD9 Komplex I wiederherstellte, die zelluläre Energie (ATP) steigerte, ROS reduzierte und die Osteoklastendifferenzierung abschwächte. Das Entfernen von ROS mit einem Antioxidans kehrte die Effekte des ACAD9-Verlusts nur teilweise um, was darauf hindeutet, dass ACAD9 auch über einen weiteren, nicht‑mitochondrialen Weg wirkt.
ACAD9 als molekularer Bremsklotz für Knochenfresser
Das Team untersuchte anschließend TRAF6, die zentrale Schaltstelle, die RANKL nutzt, um Osteoklastengene einzuschalten. Sie entdeckten, dass ein Teil des ACAD9-Proteins im Zellplasma statt in den Mitochondrien vorkommt, wo es physisch an TRAF6 binden kann. Diese Bindung verändert, wie TRAF6 mit seinen Partnerenzymen interagiert, die kleine „Ubiquitin“-Markierungen anbringen. Ohne ACAD9 wird TRAF6 stark mit einer bestimmten Art von Ubiquitin-Kette (K63-verkettet) versehen, die als Gerüst wirkt, um nachgeschaltete Signale zu aktivieren, darunter MAPK- und NF‑κB‑Wege, die Osteoklastenwachstum und Knochenresorption antreiben. In Anwesenheit von ACAD9 werden diese K63-Ketten eingeschränkt, während eine andere Art von Ubiquitin-Kette (K48-verkettet) bevorzugt wird, die TRAF6 für den Abbau in der zellulären Proteasomenmaschinerie markiert. Im Wesentlichen dämpft ACAD9 sowohl die Signalfähigkeit von TRAF6 als auch beschleunigt dessen Entfernung.
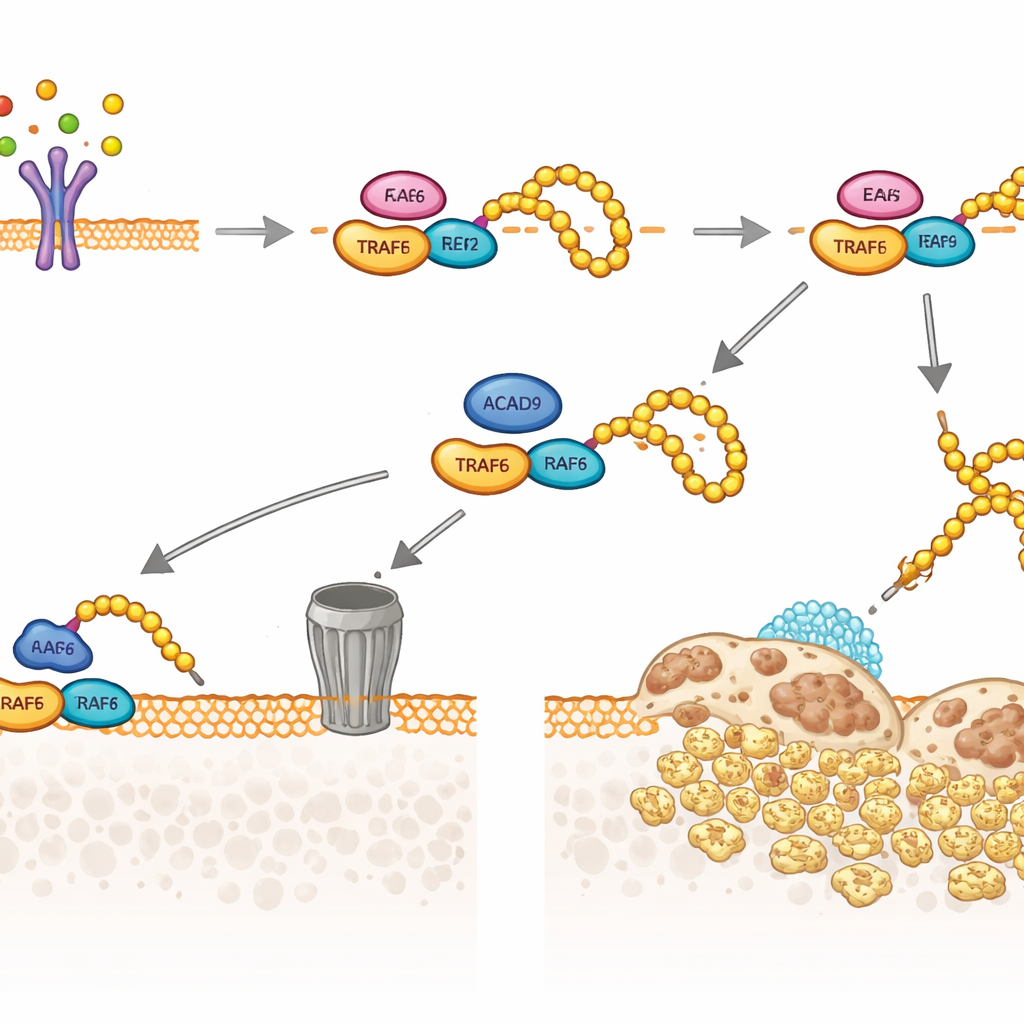
Von Maus-Knochen zu menschlichen Frakturen
Um zu sehen, wie sich das in einem lebenden Organismus auswirkt, erzeugten die Forschenden Mäuse, denen ACAD9 spezifisch in Osteoklastvorläufern fehlte. Mit zunehmendem Alter zeigten Scans dieser Tiere eine stetig dünner werdende trabekuläre Knochenstruktur, größere Abstände zwischen den Knochenbälkchen und eine schlechtere Knochenarchitektur — alles Kennzeichen von Osteoporose. Ihre Knochen wiesen mehr Osteoklasten und höhere Werte von Knochenabbaustoffwechselmarkern auf, während mitochondrialer Komplex I, Superkomplexe und ATP reduziert waren. Signalwege unterhalb von TRAF6 waren überaktiviert und ROS‑produzierende Enzyme erhöht. Wichtig war, dass die knochenaufbauende Aktivität weitgehend normal blieb, was zeigt, dass der übermäßige Knochenverlust hauptsächlich von überaktiven Osteoklasten herrührte. Ergänzend zu den Mausdaten verband eine Analyse großer humaner Biobank‑Daten seltene, schädigende Varianten im ACAD9‑Gen mit einem höheren Risiko für Wirbelfrakturen, was auf klinische Relevanz hindeutet.
Was das für künftige Therapien bedeutet
Insgesamt präsentieren die Befunde ACAD9 als zweifachen Verteidiger gegen Osteoporose. In den Mitochondrien erhält es eine effiziente Energieproduktion und begrenzt oxidativen Stress; im Zytosol bremst es TRAF6, den Hauptschalter für Osteoklastenaktivierung, indem es dessen Ubiquitin‑Markierungen umgestaltet und seinen Abbau fördert. Fehlt ACAD9 oder ist es vermindert, vermehren sich die knochenabbauenden Zellen und die Knochen werden schwächer. Wirkstoffe, die die Aktivität von ACAD9 steigern oder seine Interaktion mit TRAF6 nachahmen, könnten daher eine neue, zweigleisige Strategie bieten, um alternde Knochen zu schützen — indem sie gleichzeitig den Zellstoffwechsel verbessern und übermäßige Knochenresorption dämpfen.
Zitation: Wang, M., Yuan, C., Zhang, Y. et al. Moonlighting cytosolic function of ACAD9: suppression of TRAF6-mediated osteoclastogenesis and protection against osteoporosis. Cell Death Dis 17, 362 (2026). https://doi.org/10.1038/s41419-026-08626-z
Schlüsselwörter: Osteoporose, Osteoklasten, Mitochondrien, Ubiquitinierung, Knochenumbau