Clear Sky Science · en
Moonlighting cytosolic function of ACAD9: suppression of TRAF6-mediated osteoclastogenesis and protection against osteoporosis
Why Bones Weaken With Age
Broken hips and fragile spines are among the most feared problems of aging, yet the hidden cell biology behind these fractures is rarely discussed outside the lab. This study uncovers a surprising role for a mitochondrial protein called ACAD9 in keeping our bones strong. By acting both inside the cell’s power stations and in the cell’s signaling machinery, ACAD9 helps rein in bone‑eating cells and may open a new path for preventing osteoporosis.
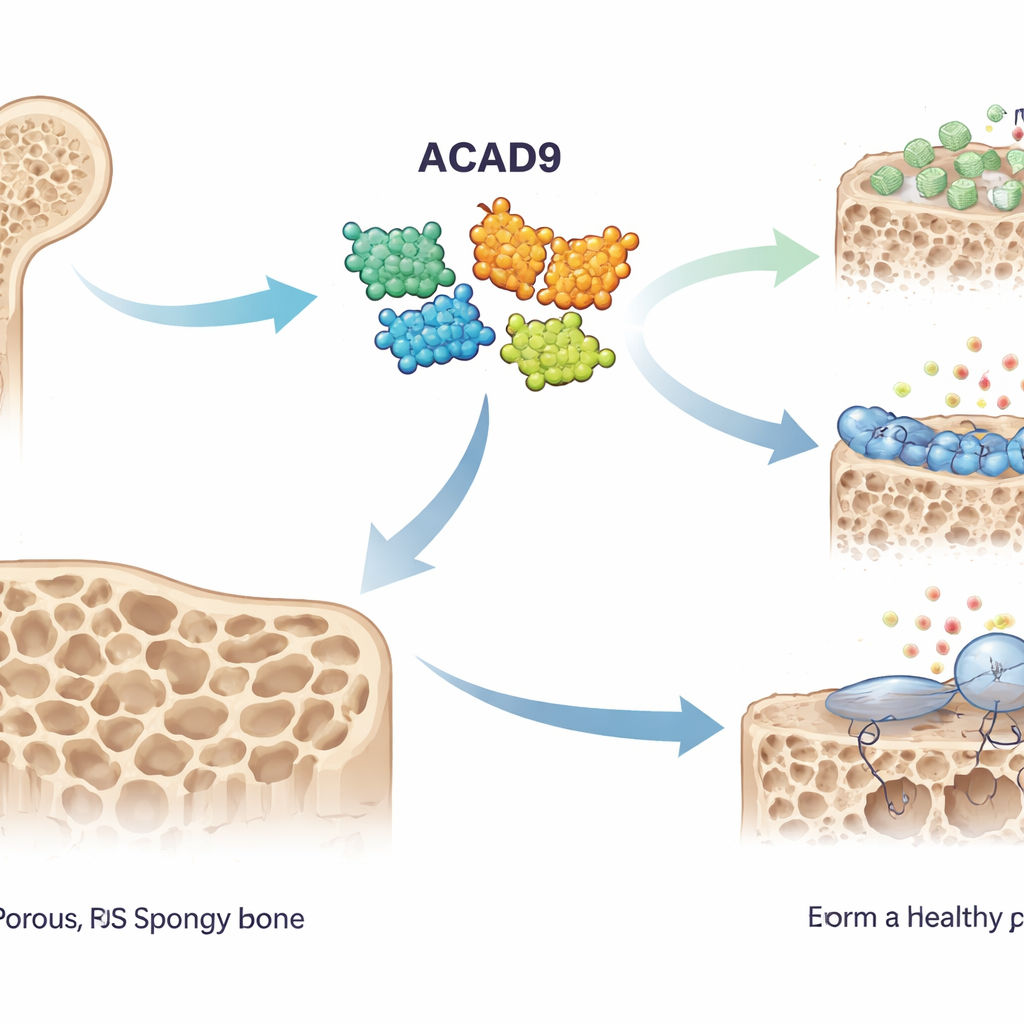
Bone Builders and Bone Eaters
Healthy bone is constantly remodeled by two opposing teams of cells: osteoblasts that build bone and osteoclasts that dissolve it. In osteoporosis, the balance tilts toward the bone eaters, leaving the skeleton thin and fragile. Osteoclasts are activated when a signal called RANKL binds to its receptor on precursor cells, switching on a protein named TRAF6. This in turn triggers chemical cascades and a burst of reactive oxygen species—highly reactive molecules often called ROS—that push these precursors to fuse into large, bone‑resorbing cells. The authors suspected that ACAD9, known for its role in mitochondrial energy production, might also influence this process.
A Mitochondrial Guardian of Stress
Inside mitochondria, ACAD9 helps assemble a large protein machine called complex I and promotes the formation of “supercomplexes,” tidy groupings of several respiratory chain complexes. These structures make energy production more efficient and limit unwanted ROS. In cell experiments, the researchers found that when osteoclast precursors were exposed to RANKL and began to mature, ACAD9 levels fell, complex I and supercomplexes declined, and ROS rose sharply. Knocking down ACAD9 further amplified ROS and sped up osteoclast formation, while boosting ACAD9 restored complex I, increased cellular energy (ATP), reduced ROS, and dampened osteoclast differentiation. Clearing ROS with an antioxidant drug only partly reversed the effects of ACAD9 loss, hinting that ACAD9 must also act through another, non‑mitochondrial route.
ACAD9 as a Molecular Brake on Bone Eaters
The team next looked at TRAF6, the key relay that RANKL uses to turn on osteoclast genes. They discovered that some ACAD9 protein resides in the cell fluid rather than in mitochondria, where it can physically bind to TRAF6. This binding changes how TRAF6 interacts with its partner enzymes that attach small “ubiquitin” tags. Without ACAD9, TRAF6 becomes heavily decorated with one type of ubiquitin chain (linked through a position called K63), which acts as a scaffold to activate downstream signals, including MAPK and NF‑κB pathways that drive osteoclast growth and bone resorption. With ACAD9 present, these K63 chains are curtailed, while a different type of ubiquitin chain (K48‑linked) is favored, targeting TRAF6 for destruction in the cell’s protein‑shredding machinery. In essence, ACAD9 both blunts TRAF6’s ability to signal and speeds its removal.
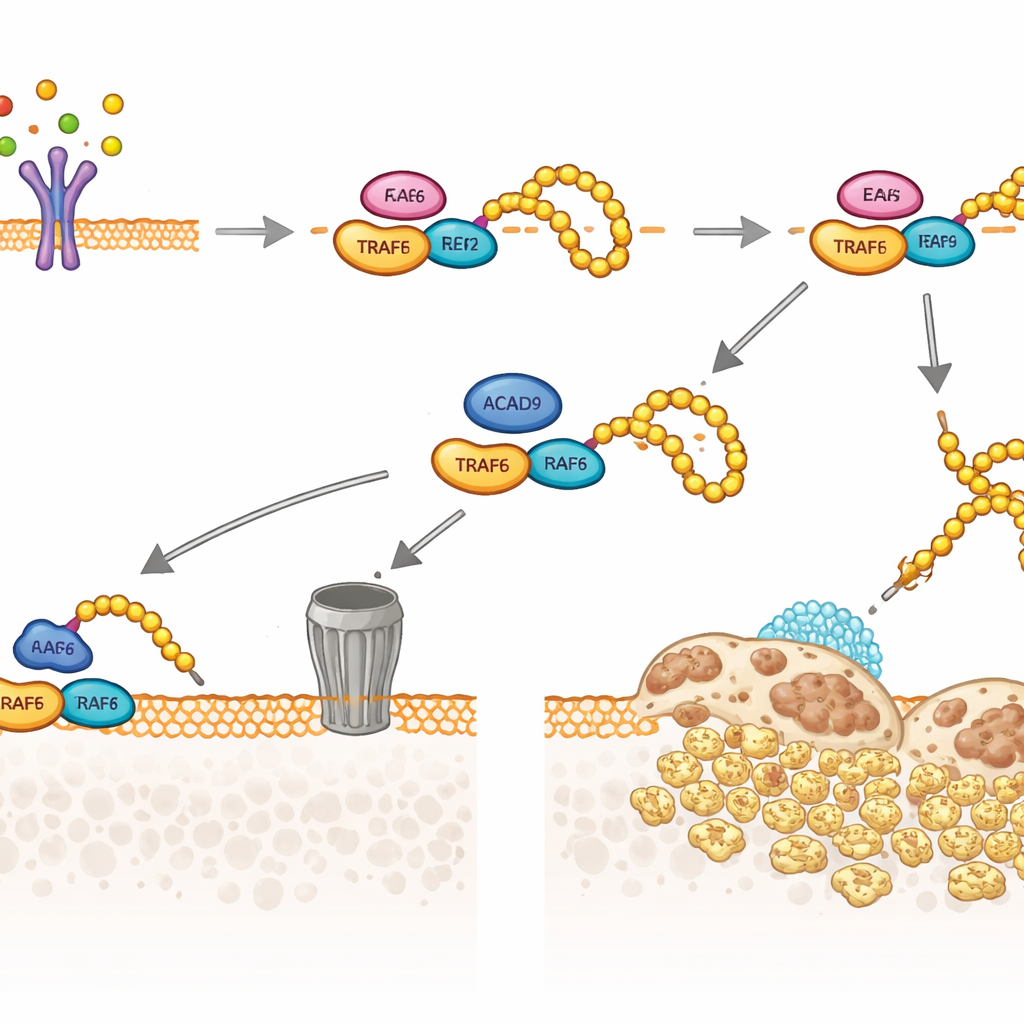
From Mouse Bones to Human Fractures
To see how this plays out in a living organism, the researchers engineered mice that lack ACAD9 specifically in osteoclast precursors. As these animals aged, scans revealed steadily thinning trabecular bone, greater spacing between bone struts, and poorer bone architecture—all hallmarks of osteoporosis. Their bones contained more osteoclasts and higher levels of bone‑resorption markers, while mitochondrial complex I, supercomplexes, and ATP were reduced. Signaling molecules downstream of TRAF6 were hyper‑activated, and ROS‑producing enzymes were elevated. Importantly, bone‑forming activity remained largely normal, showing that excess bone loss arose mainly from overactive osteoclasts. Complementing the mouse work, analysis of large human biobank data linked rare, damaging variants in the ACAD9 gene to a higher risk of vertebral fractures, suggesting clinical relevance.
What This Means for Future Therapies
Taken together, the findings present ACAD9 as a dual defender against osteoporosis. Inside mitochondria, it maintains efficient energy production and keeps oxidative stress in check; in the cytosol, it clamps down on TRAF6, the master switch for osteoclast activation, by reshaping its ubiquitin tags and promoting its breakdown. When ACAD9 is missing or reduced, bone‑eating cells multiply and bones weaken. Drugs that enhance ACAD9’s activity or mimic its interaction with TRAF6 could therefore offer a new, two‑pronged strategy to protect aging bones—simultaneously improving cellular metabolism and quieting excessive bone resorption.
Citation: Wang, M., Yuan, C., Zhang, Y. et al. Moonlighting cytosolic function of ACAD9: suppression of TRAF6-mediated osteoclastogenesis and protection against osteoporosis. Cell Death Dis 17, 362 (2026). https://doi.org/10.1038/s41419-026-08626-z
Keywords: osteoporosis, osteoclasts, mitochondria, ubiquitination, bone remodeling