Clear Sky Science · es
La composición de aminoácidos impulsa la agregación durante la síntesis de péptidos
Por qué las cadenas diminutas importan para los grandes fármacos
Muchos fármacos modernos son cadenas cortas de aminoácidos denominadas péptidos. Los químicos pueden construir estas cadenas un bloque a la vez sobre pequeñas perlas, pero el proceso a menudo se atasca porque las cadenas en crecimiento se apelotonan. Este estudio muestra que no es tanto el orden exacto de los bloques lo que controla principalmente ese apelotonamiento, sino cuántas unidades de cada tipo están presentes en conjunto. Empleando aprendizaje automático y experimentos dirigidos, los autores revelan qué aminoácidos fomentan o evitan con mayor fuerza la agregación y muestran cómo usar ese conocimiento para fabricar péptidos difíciles de sintetizar de forma mucho más eficiente.
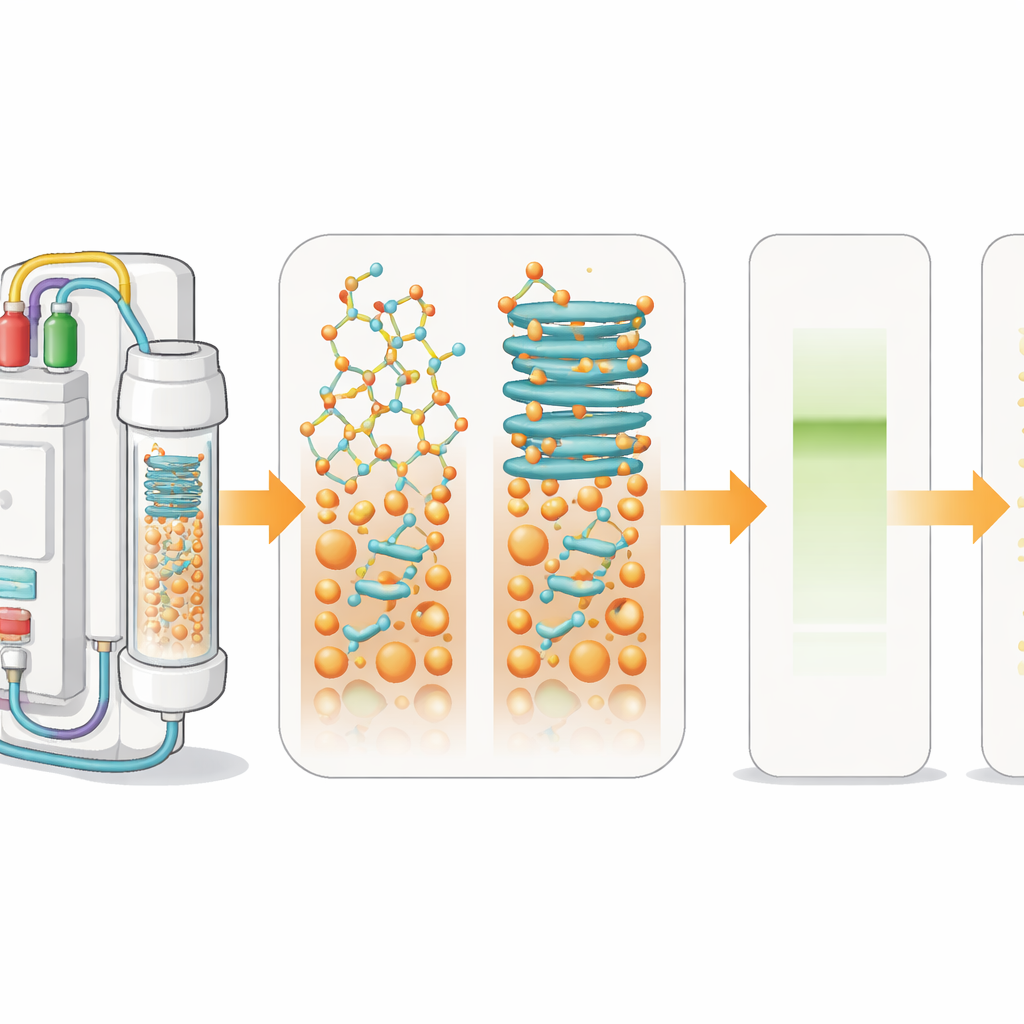
Cómo se ensamblan los fármacos péptidicos
Para fabricar péptidos en el laboratorio, los químicos usan un método llamado síntesis de péptidos en fase sólida. El primer aminoácido se ancla a una perla insoluble y luego se añaden aminoácidos adicionales en ciclos, como colocar cuentas en una pulsera. En cada paso se elimina un grupo protector y se acopla una nueva unidad. Un detector de luz integrado sigue estos pasos en tiempo real midiendo el tamaño y la forma de las señales que se generan al eliminar los grupos protectores. Cuando las cadenas en crecimiento sobre las perlas empiezan a pegarse entre sí y a plegarse en estructuras tipo hoja, las señales se ensanchan. Esta “agregación” ralentiza las reacciones y produce mezclas de productos defectuosos en lugar de un péptido único y limpio.
Dejar que los datos hablen por miles de síntesis
Los investigadores combinaron dos grandes conjuntos de datos de síntesis automatizada de péptidos en flujo, que suman más de 500 secuencias únicas. Para cada paso de cada síntesis disponían tanto del aminoácido que se añadía como de una medida cuantitativa de cuánta agregación ocurría, extraída de las señales ópticas. Aplicaron después una amplia gama de enfoques de aprendizaje automático —desde modelos modernos estilo lenguaje que leen cadenas de aminoácidos como texto, hasta modelos estadísticos clásicos y métodos de series temporales que tratan una síntesis como una secuencia de eventos— para responder una pregunta sencilla de sí o no: ¿agrega un péptido dado bajo las condiciones probadas?
Un giro sorprendente: de el orden a los ingredientes
Con todos estos métodos, la precisión de la predicción se mantuvo en un nivel modesto similar, independientemente de lo ingeniosamente que se codificara el orden de aminoácidos. Esto motivó una prueba clave. El equipo barajó aleatoriamente el orden de aminoácidos en cada péptido pero mantuvo los recuentos totales de cada tipo iguales. Si el orden preciso fuera crucial, el rendimiento debería haber colapsado. No fue así: los modelos funcionaron igual de bien con las secuencias barajadas. Incluso cuando cada péptido se redujo a un “vector de composición” de 20 números que registra simplemente la fracción de cada aminoácido presente, la precisión se mantuvo parecida. Para confirmar esto experimentalmente, los autores tomaron ocho péptidos conocidos —cuatro que agregan y cuatro que no— y sintetizaron cinco versiones barajadas de cada uno. La mayoría de las variantes barajadas se comportaron como los originales: las secuencias que agregaban siguieron agregando, las que no agregaban permanecieron mayormente solubles y tendieron a comenzar a agregarse en posiciones similares a lo largo de la cadena.
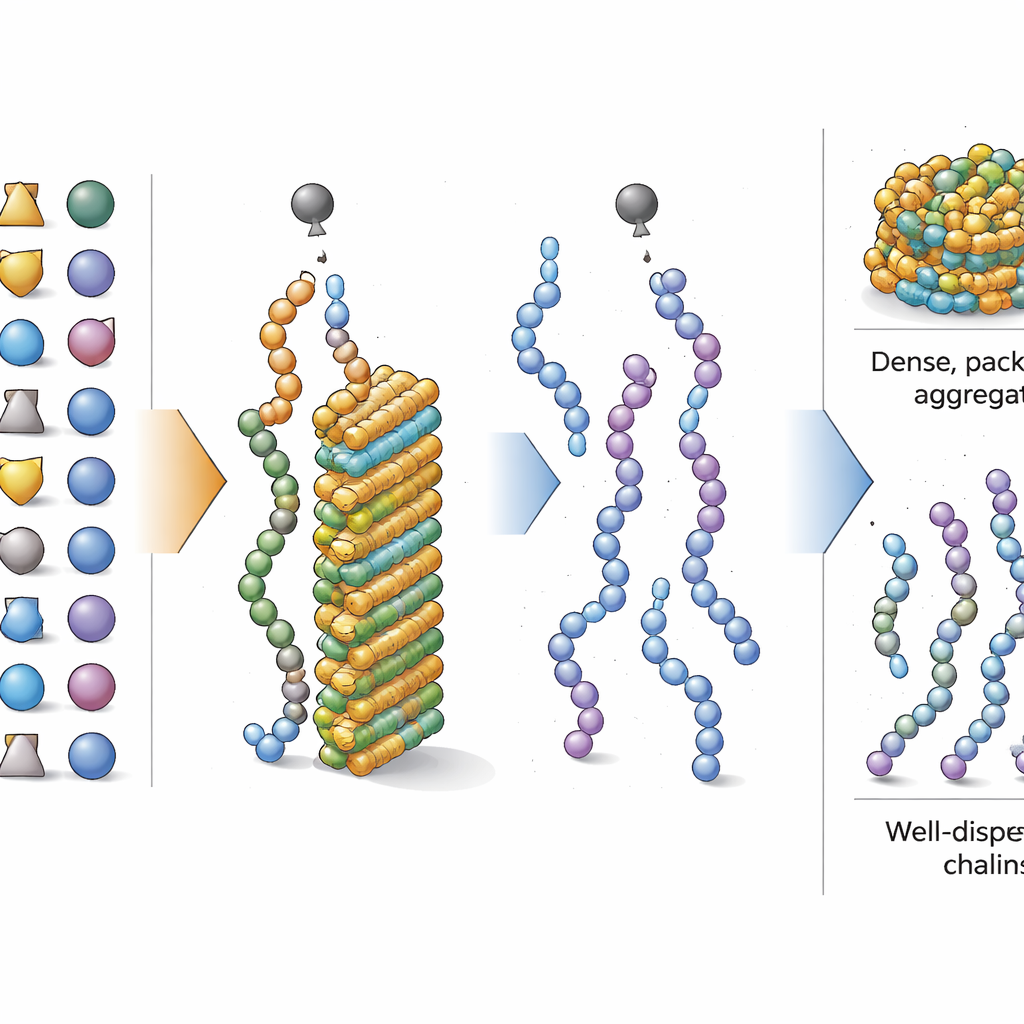
Actores buenos y malos entre los aminoácidos
Con el foco puesto en la composición global, los investigadores preguntaron a continuación qué aminoácidos inclinan más fuertemente un péptido hacia la agregación o hacia evitarla durante la síntesis. Utilizando una herramienta interpretable de aprendizaje automático, puntuaron cuánto cambiaba la predicción del modelo al aumentar la fracción de cada aminoácido. Encontraron que los bloques con cadenas laterales simples y grasas —como isoleucina y valina, así como formas protegidas comúnmente usadas de serina y treonina— promueven fuertemente la agregación cuando están presentes en altas cantidades. En contraste, aminoácidos con cadenas laterales más voluminosas o complejas, incluidas formas protegidas de fenilalanina, tirosina, ácido aspártico y arginina, tendieron a reducir la agregación. Ciertos patrones cortos de dos aminoácidos, como glicina–serina y leucina–leucina, también mostraron una tendencia añadida a causar apelotonamiento, pero el equilibrio general de tipos de aminoácidos siguió siendo el factor dominante.
Convertir los conocimientos en mejores recetas de síntesis
Armado con este mapa de aminoácidos “que apelotonan” y “que despejan”, el equipo construyó un conjunto de modelos que pueden sugerir correcciones prácticas antes de ejecutar una síntesis. Para un péptido propuesto, el programa predice si es probable que ocurra agregación y señala posiciones cerca del extremo anclado de la cadena donde los aminoácidos problemáticos contribuyen más. Luego recomienda intercambiar estos por versiones especiales, como bloques de construcción tipo pseudoprolina, que interrumpen temporalmente la agregación pero vuelven a las aminoácidos deseados tras el paso final de desprotección. Al probar esta estrategia en dos fragmentos peptídicos notoriamente problemáticos, los investigadores introdujeron un pequeño número de esas sustituciones en las posiciones seleccionadas por el modelo. El resultado fue un salto espectacular en la pureza cruda —de aproximadamente una quinta parte a cerca de tres cuartas partes de la mezcla de producto— acorde con la clasificación del modelo sobre qué posiciones ayudarían más.
Qué implica esto para los futuros fármacos péptidicos
Para un no especialista, el mensaje central es que, bajo estas condiciones sintéticas, el apelotonamiento de péptidos se comporta menos como un patrón delicado de llave y cerradura y más como un problema de receta a granel: las proporciones de los ingredientes “grasos” y “voluminosos” importan más que su orden exacto. Al reconocer y cuantificar esto, los autores pudieron tanto explicar fracasos de síntesis persistentes como ofrecer una forma sencilla y basada en datos para evitarlos. Su trabajo demuestra cómo el aprendizaje automático puede descubrir reglas ocultas en procesos químicos y convertirlas en herramientas de diseño directas, acelerando finalmente el camino desde ideas peptídicas hasta fármacos fiables y escalables.
Cita: Tamás, B., Alberts, M., Laino, T. et al. Amino acid composition drives aggregation during peptide synthesis. Nat. Chem. 18, 677–685 (2026). https://doi.org/10.1038/s41557-026-02090-0
Palabras clave: síntesis de péptidos, composición de aminoácidos, agregación, aprendizaje automático, química en fase sólida