Clear Sky Science · en
Amino acid composition drives aggregation during peptide synthesis
Why tiny chains matter for big medicines
Many modern medicines are short chains of amino acids called peptides. Chemists can build these chains one building block at a time on tiny beads, but the process often stalls because the growing chains clump together. This study shows that it is not the exact order of building blocks that mainly controls this clumping, but how many of each type are present overall. Using machine learning and targeted experiments, the authors reveal which amino acids most strongly drive or prevent clumping and show how to use this knowledge to make hard-to-synthesize peptides much more efficiently.
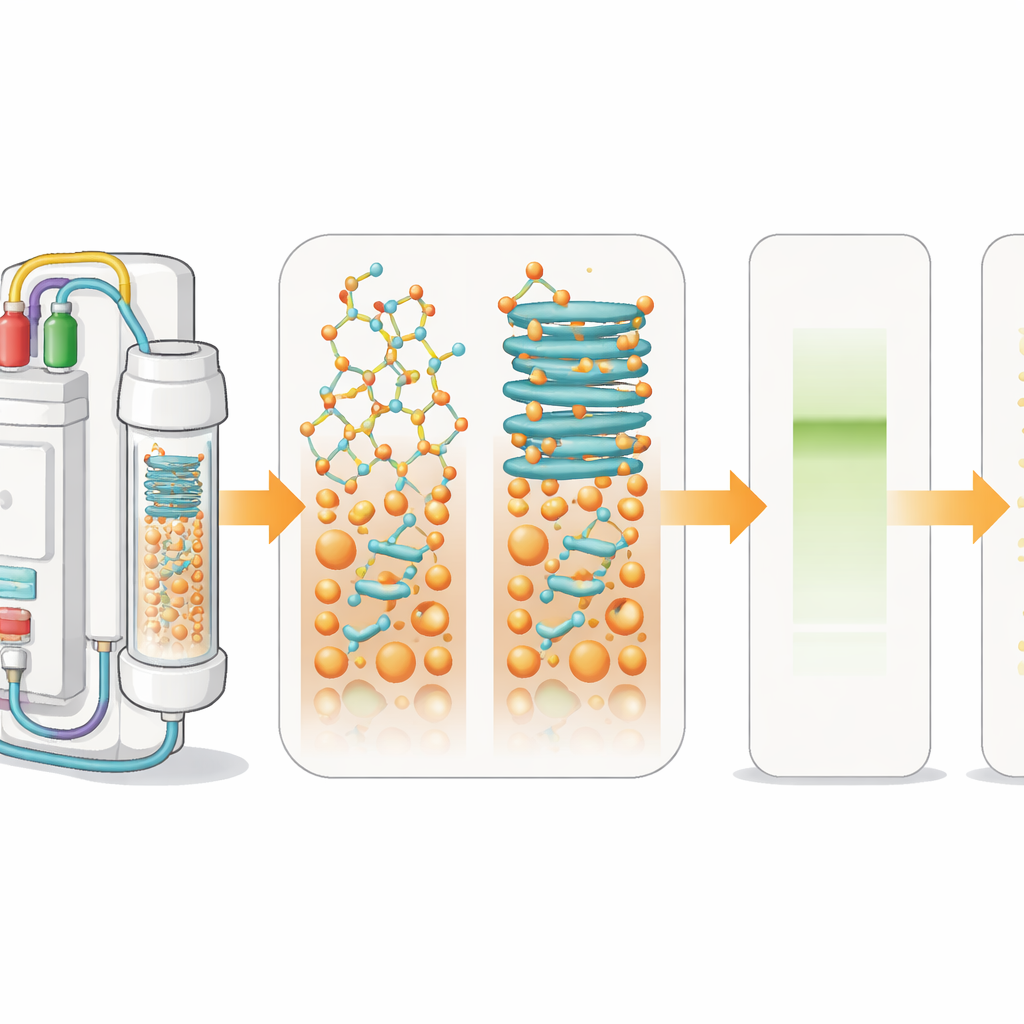
How peptide drugs are stitched together
To make peptides in the lab, chemists use a method called solid-phase peptide synthesis. The first amino acid is attached to an insoluble bead, and then additional amino acids are added in cycles, like snapping beads onto a bracelet. During each step, a protective group is removed and a new unit is coupled. A built-in light detector tracks these steps in real time by measuring the size and shape of signals produced when protection groups are removed. When the growing chains on the beads begin to stick together and fold into sheet-like structures, the signals broaden. This “aggregation” slows down reactions and leads to mixtures of faulty products instead of a single clean peptide.
Letting data speak for thousands of syntheses
The researchers combined two large datasets of automated flow-based peptide syntheses, totaling more than 500 unique sequences. For each step in each synthesis, they had both the amino acid being added and a quantitative measure of how much aggregation was occurring, extracted from the light-based signals. They then applied a wide range of machine learning approaches—from modern language-style models that read amino acid strings like text, to classic statistical models and time-series methods that treat a synthesis as a sequence of events—to answer a simple yes–no question: does a given peptide aggregate under the tested conditions?
A surprising shift from order to ingredients
Across all these methods, the prediction accuracy hovered around the same modest level, regardless of how cleverly the amino acid order was encoded. This prompted a key test. The team randomly shuffled the order of amino acids in each peptide but kept the overall counts of each type the same. If precise order were crucial, performance should have collapsed. It did not: the models did just as well on shuffled sequences. Even when each peptide was reduced to a 20-number “composition vector” that simply records the fraction of each amino acid present, accuracy stayed similar. To confirm this experimentally, the authors took eight known peptides—four that aggregate and four that do not—and synthesized five shuffled versions of each. Most shuffled variants behaved like the originals: aggregating sequences still aggregated, and non-aggregating ones largely stayed soluble, and they tended to start aggregating at similar positions along the chain.
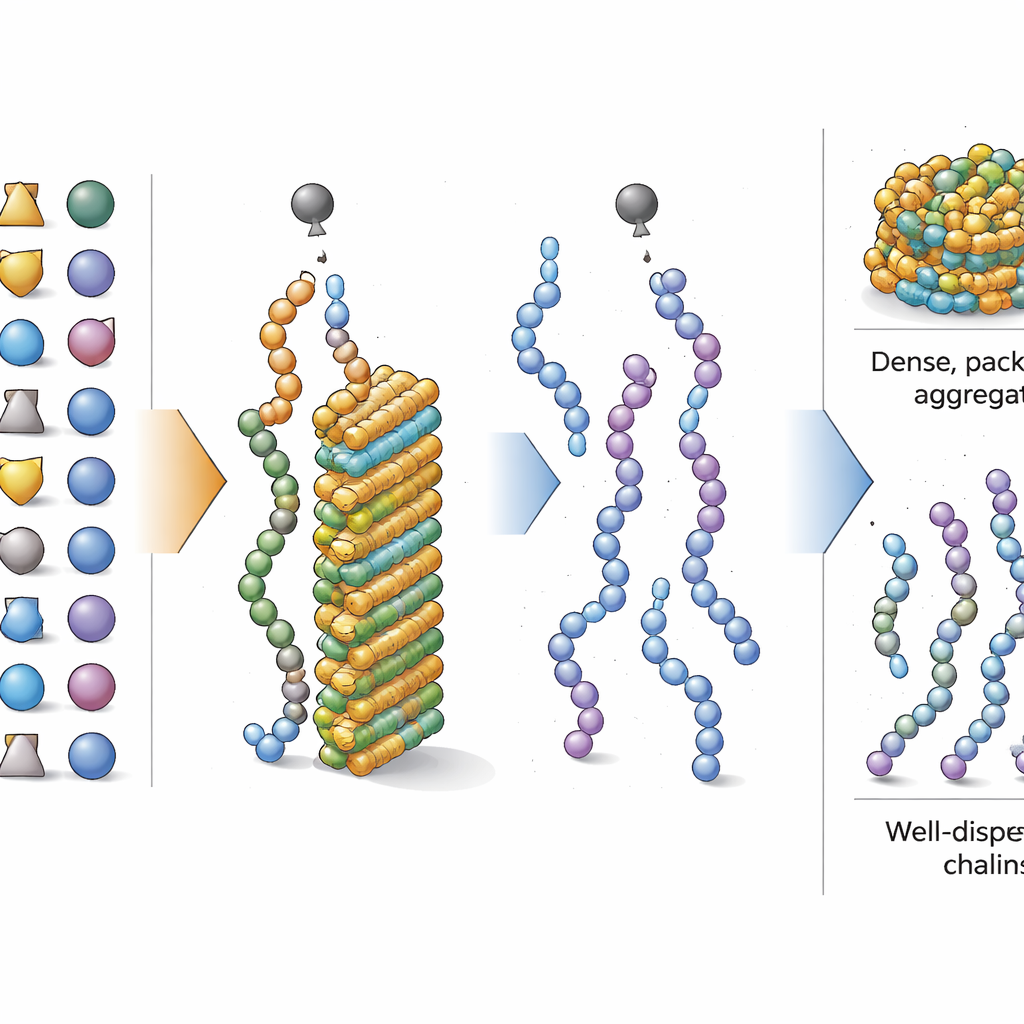
Good and bad actors among amino acids
With the focus shifted to overall composition, the researchers next asked which amino acids most strongly tip a peptide toward or away from aggregation during synthesis. Using an interpretable machine learning tool, they scored how much increasing the fraction of each amino acid changed the model’s prediction. They found that building blocks with simple, oily side chains—such as isoleucine and valine, as well as commonly used protected forms of serine and threonine—strongly promote aggregation when present in high amounts. By contrast, amino acids with bulkier or more complex side groups, including protected forms of phenylalanine, tyrosine, aspartic acid and arginine, tended to reduce aggregation. Certain short two-amino-acid patterns, like glycine–serine and leucine–leucine, also showed added tendencies to cause clumping, but the overall balance of amino acid types remained the dominant factor.
Turning insights into better synthesis recipes
Armed with this map of “clumping” and “declumping” amino acids, the team built an ensemble of models that can suggest practical fixes before a synthesis is run. For a proposed peptide, the program predicts whether aggregation is likely and pinpoints positions near the anchored end of the chain where problem amino acids contribute most. It then recommends swapping these for special versions, such as pseudoproline building blocks, that temporarily disrupt aggregation but revert to the desired amino acids after the final deprotection step. Testing this strategy on two notoriously troublesome peptide fragments, the researchers introduced a small number of such substitutions at model-selected positions. The result was a dramatic jump in crude purity—from roughly one-fifth to about three-quarters of the product mixture—in line with the model’s ranking of which positions would help most.
What this means for future peptide medicines
To a non-specialist, the central message is that, for these synthetic conditions, peptide clumping behaves less like a delicate lock-and-key pattern and more like a bulk recipe problem: the proportions of the “oily” and “bulky” ingredients matter more than their exact order. By recognizing and quantifying this, the authors could both explain longstanding synthesis failures and offer a simple, data-driven way to avoid them. Their work showcases how machine learning can uncover hidden rules in chemical processes and convert them into straightforward design tools, ultimately speeding the path from peptide ideas to reliable, scalable medicines.
Citation: Tamás, B., Alberts, M., Laino, T. et al. Amino acid composition drives aggregation during peptide synthesis. Nat. Chem. 18, 677–685 (2026). https://doi.org/10.1038/s41557-026-02090-0
Keywords: peptide synthesis, amino acid composition, aggregation, machine learning, solid-phase chemistry