Clear Sky Science · en
Accelerating natural product discovery with linked MS-genomics and language/transformer-based models
Why new medicines hide in plain sight
Many of our most important medicines, including antibiotics and cancer drugs, come from tiny microbes that make complex natural chemicals. Yet finding new molecules in this hidden pharmacy is slow and expensive, because scientists must sift through huge libraries of microbes and guess which ones are worth testing. This paper shows how combining advanced artificial intelligence with modern lab measurements can greatly speed up the hunt, helping researchers zero in on the most promising microbes before doing laborious experiments. 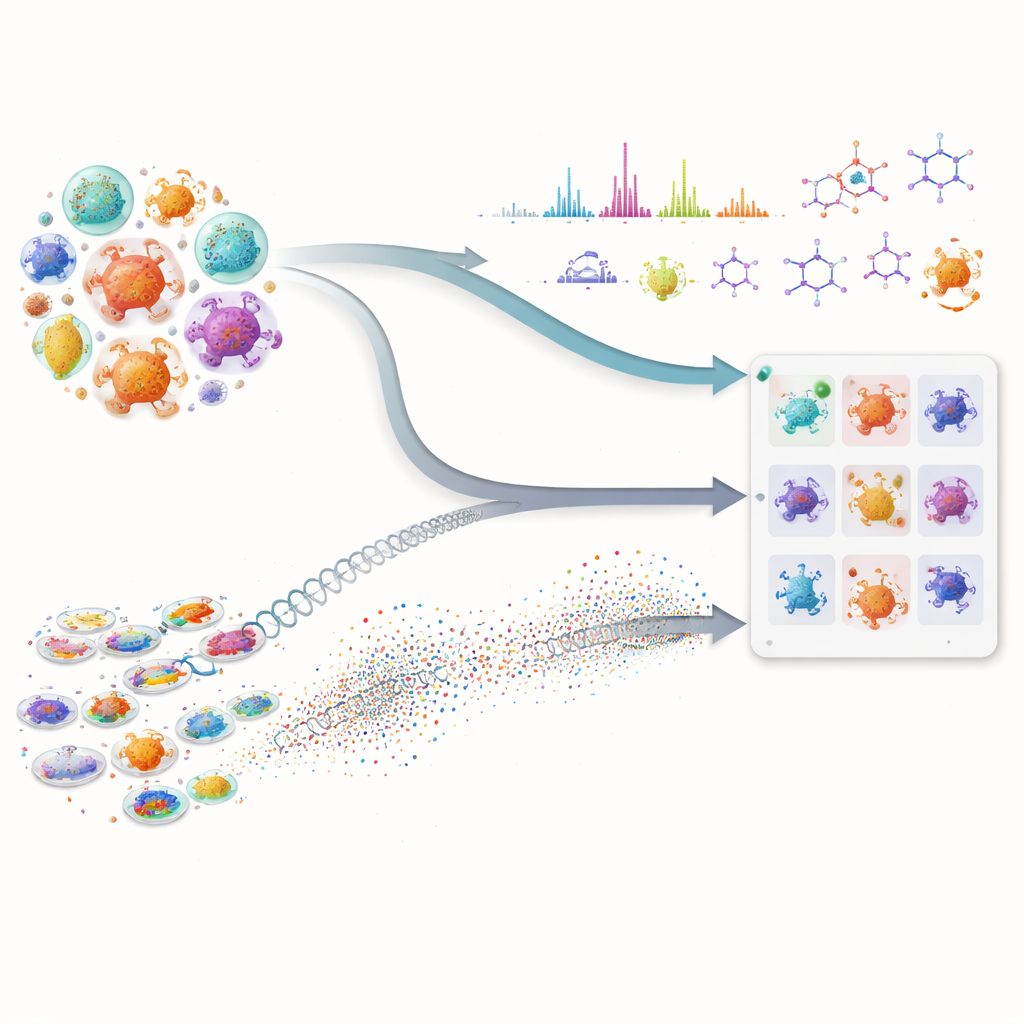
Turning microbe DNA into a searchable map
The first part of the approach looks at the genetic “blueprints” of microbes. Instead of using only traditional DNA matching, the team applies powerful protein language models—AI systems that have learned patterns across millions of protein sequences. These models convert each biosynthetic protein into a high-dimensional numerical fingerprint. That lets scientists detect distant functional cousins even when the underlying DNA has changed a lot or is split across fragments, a common problem with rough draft genomes. The researchers then score each microbe by how many of its proteins resemble those in a known pathway for making a target molecule, and by how strong that overall similarity is. This composite score highlights strains whose combined protein set looks like it could build the desired compound, while discounting microbes that only have one or two common, non-specialized enzymes.
Reading complex chemical mixtures with AI
The second part focuses on what the microbes actually produce in the lab. Using liquid chromatography and tandem mass spectrometry, scientists record detailed “fingerprints” of the molecules in fermentation broths. The authors’ in-house Workflow for Intelligent Structural Elucidation, or WISE, cleans up these signals, separates overlapping peaks, and then uses AI models trained on millions of natural product-like structures to guess which molecular shapes best match each spectrum. A transformer-based model predicts what the spectra of candidate molecules should look like, and a combined score weighs how well the observed and predicted patterns align, including fine details like isotopic patterns and exact mass. By analyzing how these scores are distributed on a standard benchmark, the team identifies thresholds that separate likely real matches from noise or look-alike decoys, allowing them to label some structural guesses as high confidence and to filter out obviously wrong ones.
Bringing genes and chemicals together
The real power of the method comes from merging these two streams of information. For any molecule of interest, a microbe rises to the top of the list only if it shows both genetic potential—its proteins resemble those in a known pathway—and chemical evidence—it produces spectral features that fit the expected structure. This cross-checking reduces false leads that appear convincing in only one type of data. The authors tested their framework on a large collection of actinobacterial strains and mutants, focusing on three very different antimicrobial compounds: valinomycin, surfactin, and the antibiotic neomycin B. Instead of relying on perfect genomes or complete spectral libraries, the system works well even when data are messy or incomplete, a common reality in early-stage discovery campaigns. 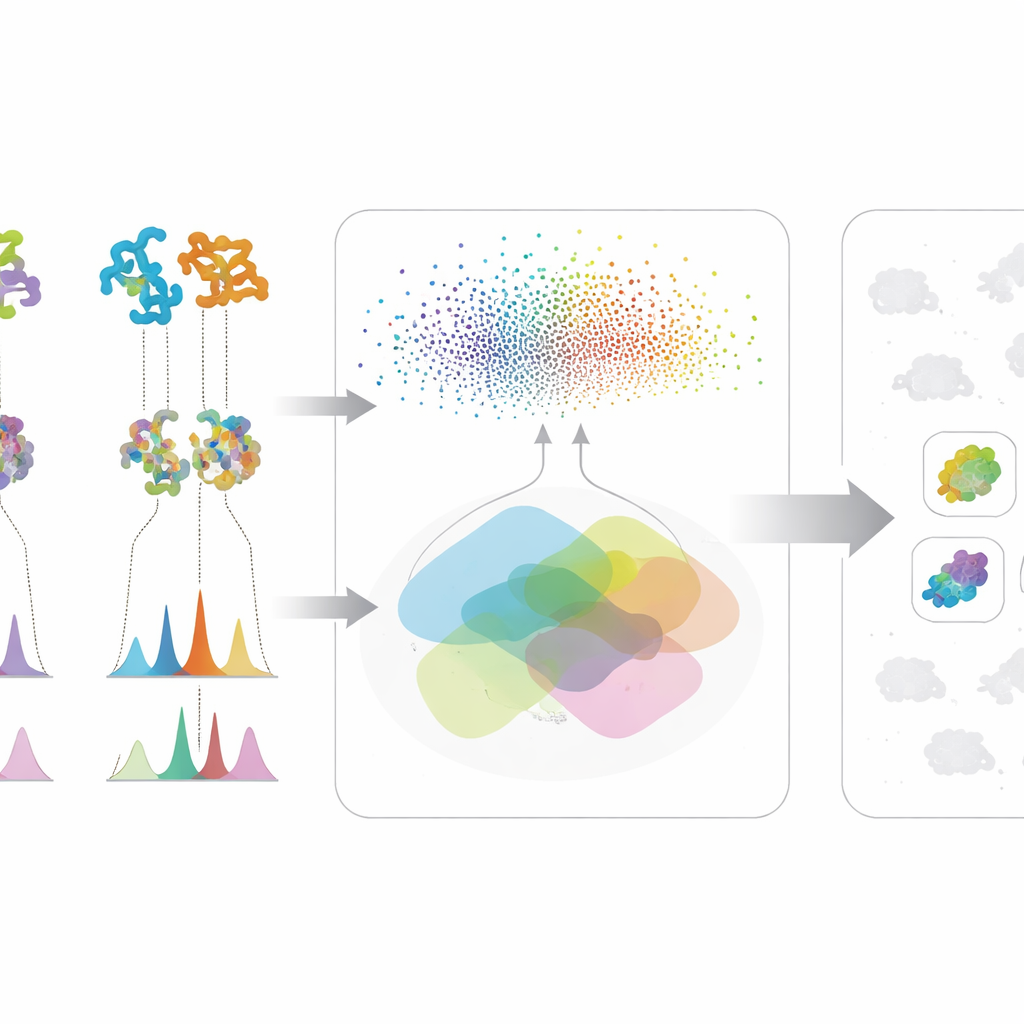
Putting the framework to the test
In the neomycin B case study, the AI first searched for microbes whose proteins looked like those in the known neomycin pathway, then checked whether their mass spectra hinted at the presence of neomycin-like molecules. Four strains passed both filters; three of them were confirmed experimentally to make neomycin B, including two previously unrecognized producers. For valinomycin and surfactin, the framework also pinpointed producers with high precision, while a control test that randomly scrambled the scores performed much worse. These results show that the model is capturing real biological relationships, not just random coincidences in the data, and that it can successfully guide researchers toward the most likely hits in a crowded search space.
What this means for future drug discovery
In everyday terms, the authors have built a smart recommendation engine for natural product discovery. Instead of testing every microbe and every chemical signal in a brute-force way, scientists can now focus on a short list of strains where genetic potential and chemical output agree. This greatly cuts down on wasted effort, while still leaving room to discover unexpected molecules that are not yet in any reference book. As AI models and datasets continue to improve, this kind of integrated genomic and metabolomic reasoning could unlock vast regions of microbial chemistry that remain unexplored, potentially revealing new antibiotics and other useful compounds just when society needs them most.
Citation: Tay, D.W.P., Koh, W., Ang, S.J. et al. Accelerating natural product discovery with linked MS-genomics and language/transformer-based models. npj Antimicrob Resist 4, 31 (2026). https://doi.org/10.1038/s44259-026-00206-7
Keywords: natural product discovery, microbial metabolites, mass spectrometry, protein language models, AI in drug discovery