Clear Sky Science · en
Lipid asymmetry disruption by XKR8 orchestrates neutrophil extracellular trap formation and inhibits fungal infection
How our first-line defenders spring a hidden trap
When germs invade, one of the body’s fastest responders is the neutrophil, a white blood cell that can throw out sticky webs of DNA to ensnare microbes. These structures, called neutrophil extracellular traps, or NETs, are powerful allies in fighting infection but can also damage our own tissues when unleashed at the wrong time. This study uncovers a surprising “master switch” in the neutrophil’s outer membrane that helps decide when these DNA webs are cast, and shows how that switch protects the lungs from dangerous fungal invaders.
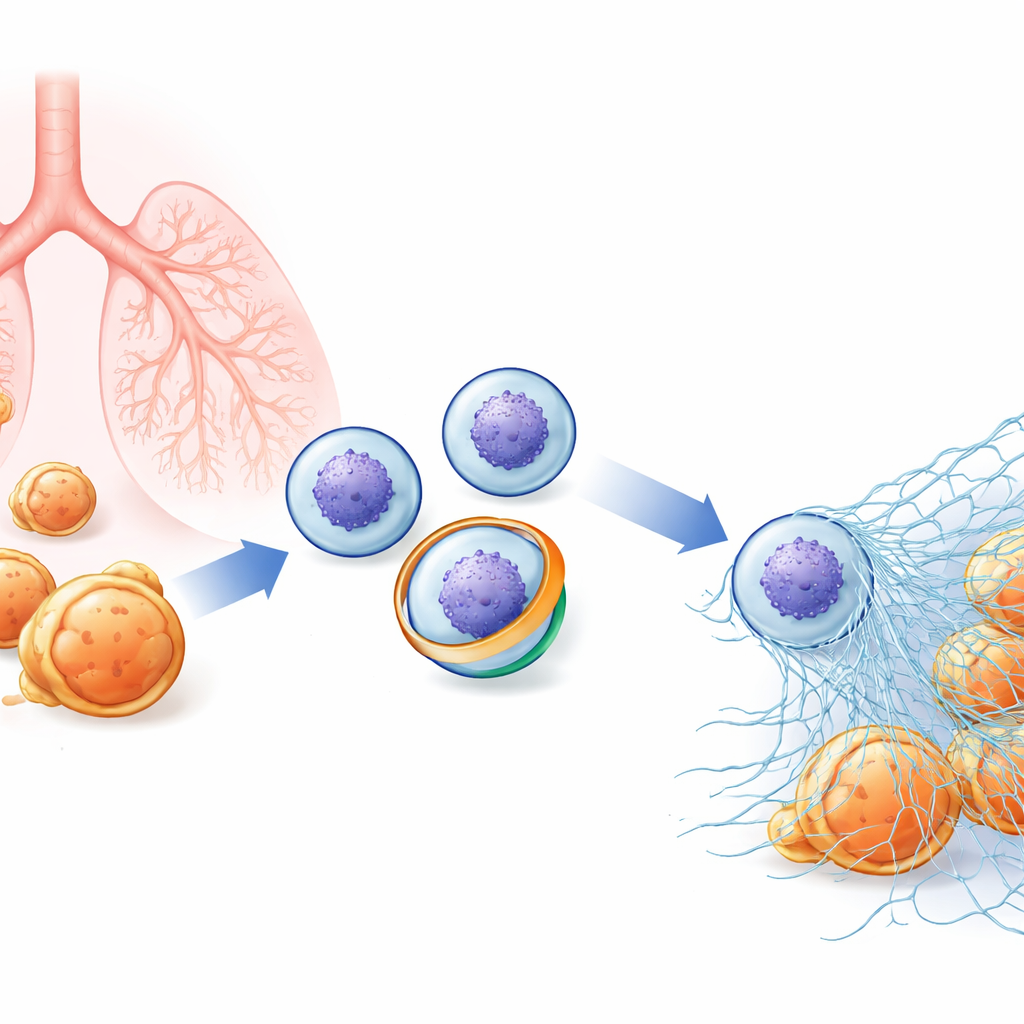
A hidden web inside immune cells
Neutrophils patrol the blood ready to attack bacteria and fungi. Besides swallowing microbes, they can explode outward, releasing decondensed chromatin (the material that makes up chromosomes) coated with antimicrobial enzymes. These sticky fibers form NETs that tangle up pathogens so they can be killed locally. Because NETs are so potent, their release has to be tightly controlled: too little, and infections can spiral out of control; too much, and the same webs can fuel autoimmune disease, lung injury, and blood clots. Researchers suspected that subtle changes in the neutrophil’s outer membrane might be an early step in deciding whether a cell will undergo this dramatic “NETosis,” but the key molecular players were unknown.
A membrane switch called XKR8
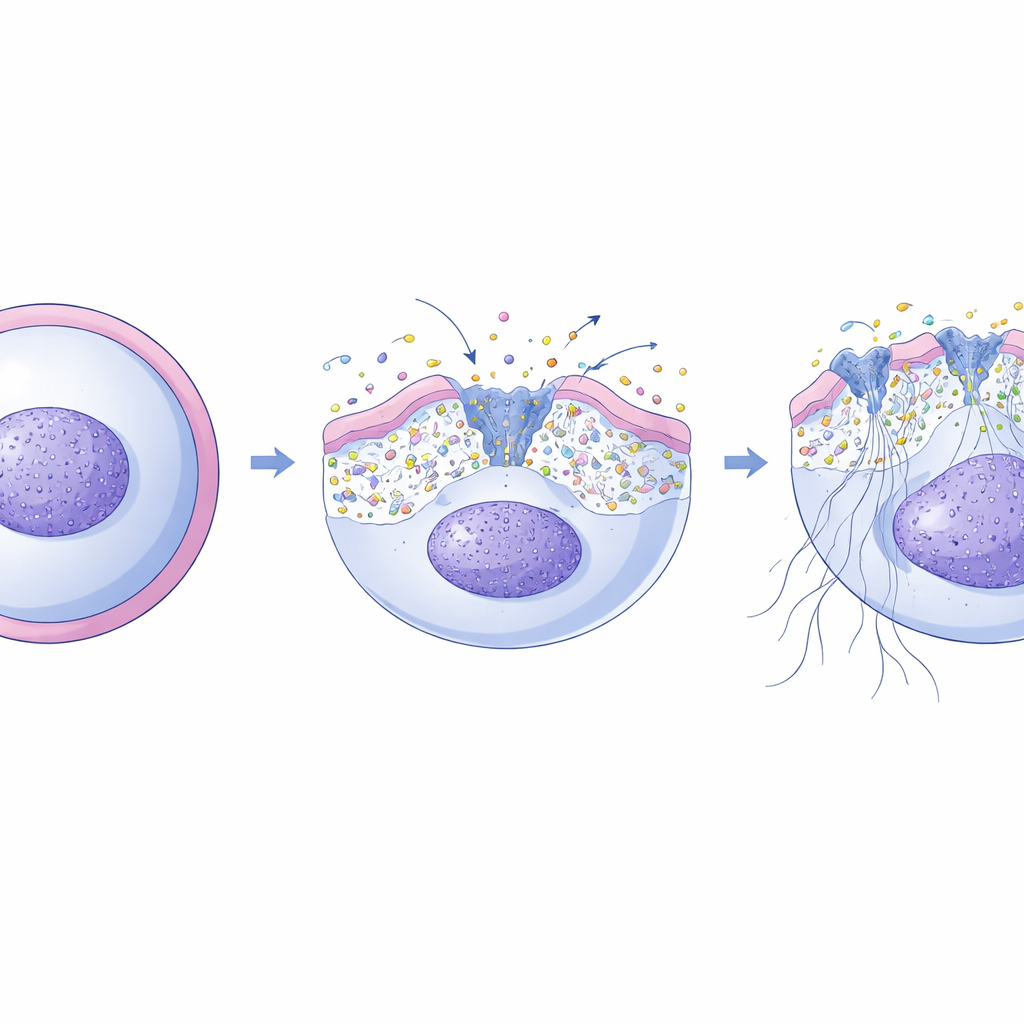
The team focused on a protein named XKR8, found at especially high levels in neutrophils. XKR8 is a “scramblase” – a protein that can flip lipids between the inner and outer layers of the cell membrane, erasing the usual asymmetry between the two sides. In other contexts, such as programmed cell death, XKR8 is known to expose a lipid signal on the cell surface that marks dying cells for removal. Here, the authors showed that during NET formation, exposure of this lipid signal appears before the membrane becomes leaky and before DNA is released, marking those neutrophils that are destined to form NETs. When they removed XKR8 from mouse and human neutrophils, the cells could no longer carry out this early scrambling step and almost completely failed to make NETs in response to many chemical and microbial triggers.
From membrane reshuffle to calcium surge
NET formation is known to depend on a rise in calcium inside neutrophils, which activates enzymes that loosen chromatin and allow DNA to spill out. The researchers discovered that XKR8 sits at the heart of this process. After an oxidative burst inside the neutrophil, an executioner enzyme called caspase-3 cleaves and activates XKR8. Once switched on, XKR8 rapidly redistributes several types of lipids across the membrane, not just a single “eat me” signal. Using fluorescent probes, the team showed that this reshuffling changes the physical tension of the membrane. In turn, this mechanical stress opens up a set of mechanosensitive calcium channels embedded in the membrane, allowing calcium ions to stream into the cell precisely during the window when NET formation is underway. If the researchers blocked these channels, NETs were largely abolished; if they stimulated the channels with special activators, they could restore NET formation even in cells lacking XKR8.
When traps fail: lung injury and fungal infection
To understand what this membrane switch means in living animals, the team studied mice engineered either to lack XKR8 entirely or to carry a mutated version that cannot be cut by caspase-3. In models of acute lung injury triggered by bacterial components, normal mice developed abundant NETs in their lungs, high levels of free DNA in lung fluid, and marked tissue damage. Mice with impaired XKR8 activity recruited the same number of neutrophils but produced far fewer NETs and suffered less lung injury, suggesting that the XKR8 switch is a major driver of harmful NET release in this setting. In contrast, when the lungs were infected with the fungus Candida albicans, mice without a working XKR8 switch were at a serious disadvantage: they formed far fewer NETs, carried heavier fungal burdens in their lungs and other organs, bled more into the air spaces, and died more often from overwhelming infection. Notably, a drug that activates one of the mechanosensitive calcium channels partly rescued fungal control in mice whose neutrophils lacked XKR8.
One central lever with two faces
This work reveals a unifying mechanism by which many different danger signals converge on a single membrane-based decision point in neutrophils. Oxidative bursts lead to caspase-3 activation, which flips on XKR8 to scramble membrane lipids, change membrane tension, and open calcium channels, culminating in the launch of NETs. For a lay reader, the message is that our immune system uses a finely tuned “lipid lever” on the surface of neutrophils to decide when to cast DNA webs. Tipping that lever one way helps clear large fungal invaders; tipping it too often or at the wrong time can worsen sterile inflammation such as lung injury or arthritis. Because this pathway sits at the crossroads of protection and damage, targeting the caspase-3–XKR8–calcium axis could provide new strategies to either rein in harmful NETs or boost beneficial ones, depending on the clinical need.
Citation: Liu, W., Ping, J., Deng, L. et al. Lipid asymmetry disruption by XKR8 orchestrates neutrophil extracellular trap formation and inhibits fungal infection. Nat Immunol 27, 949–960 (2026). https://doi.org/10.1038/s41590-026-02456-z
Keywords: neutrophil extracellular traps, XKR8 scramblase, innate immunity, fungal lung infection, acute lung injury