Clear Sky Science · de
Störung der Lipid-Asymmetrie durch XKR8 steuert die Bildung neutrophiler extrazellulärer Netze und hemmt Pilzinfektionen
Wie unsere ersten Verteidiger eine verborgene Falle auswerfen
Wenn Krankheitserreger eindringen, ist einer der schnellsten Helfer des Körpers das Neutrophil, eine weiße Blutzelle, die klebrige DNA-Netze auswerfen kann, um Mikroben zu fangen. Diese Strukturen, neutrophile extrazelluläre Netze oder NETs genannt, sind starke Verbündete im Kampf gegen Infektionen, können aber auch eigenes Gewebe schädigen, wenn sie zur falschen Zeit freigesetzt werden. Die Studie deckt einen überraschenden „Hauptschalter“ in der Außenmembran von Neutrophilen auf, der mitentscheidet, wann diese DNA-Netze ausgeworfen werden, und zeigt, wie dieser Schalter die Lunge vor gefährlichen Pilzangreifern schützt.
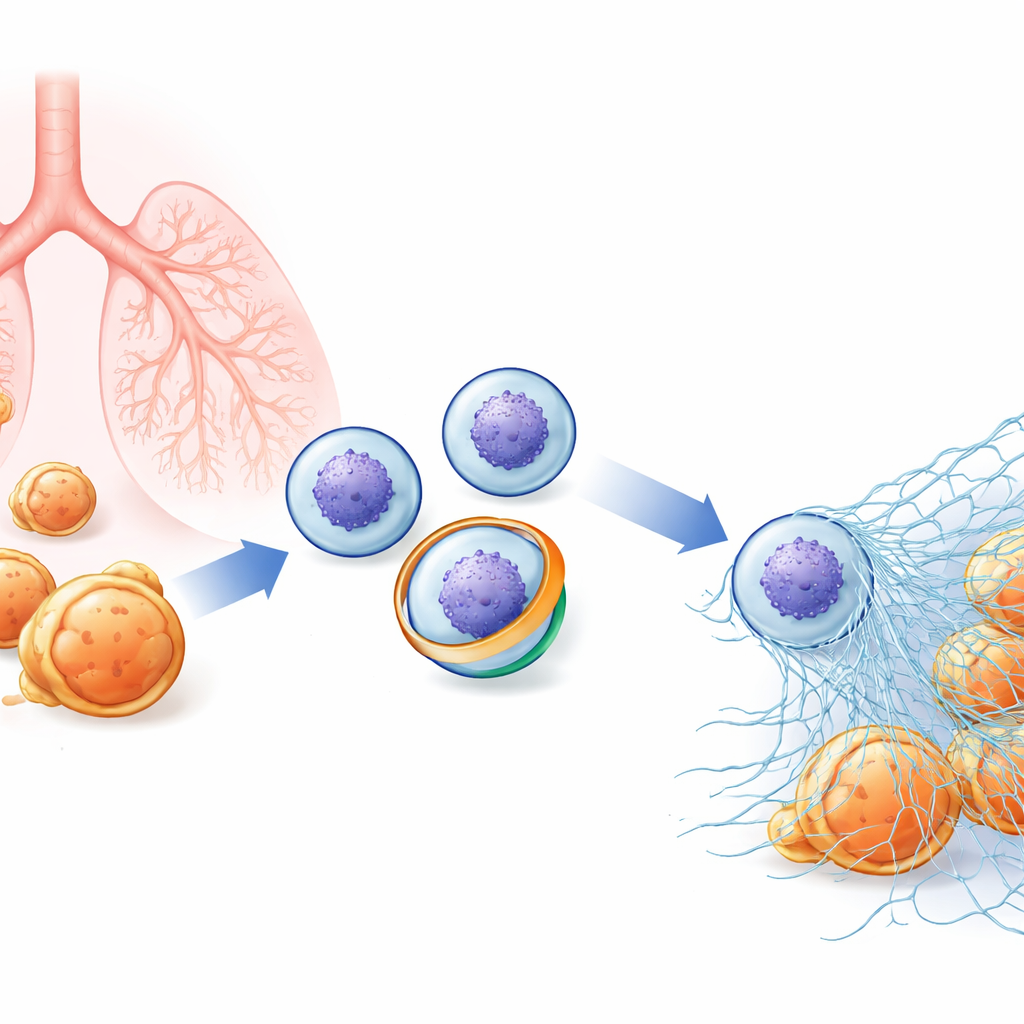
Ein verborgenes Netz in Immunzellen
Neutrophile patrouillieren im Blut und sind bereit, Bakterien und Pilze anzugreifen. Neben dem Verschlucken von Mikroben können sie nach außen aufplatzen und dekondensiertes Chromatin (das Material, aus dem Chromosomen bestehen) freisetzen, das mit antimikrobiellen Enzymen überzogen ist. Diese klebrigen Fasern bilden NETs, die Pathogene verfangen, damit sie lokal getötet werden können. Da NETs so wirkmächtig sind, muss ihre Freisetzung streng reguliert sein: Zu wenig führt zu unkontrollierten Infektionen; zu viel kann Autoimmunerkrankungen, Lungenschäden und Blutgerinnsel fördern. Forscher vermuteten, dass subtile Veränderungen in der Außenmembran von Neutrophilen ein frühes Signal dafür sein könnten, ob eine Zelle diese dramatische „NETose“ durchläuft, doch die entscheidenden molekularen Akteure waren bisher unbekannt.
Ein Membranschalter namens XKR8
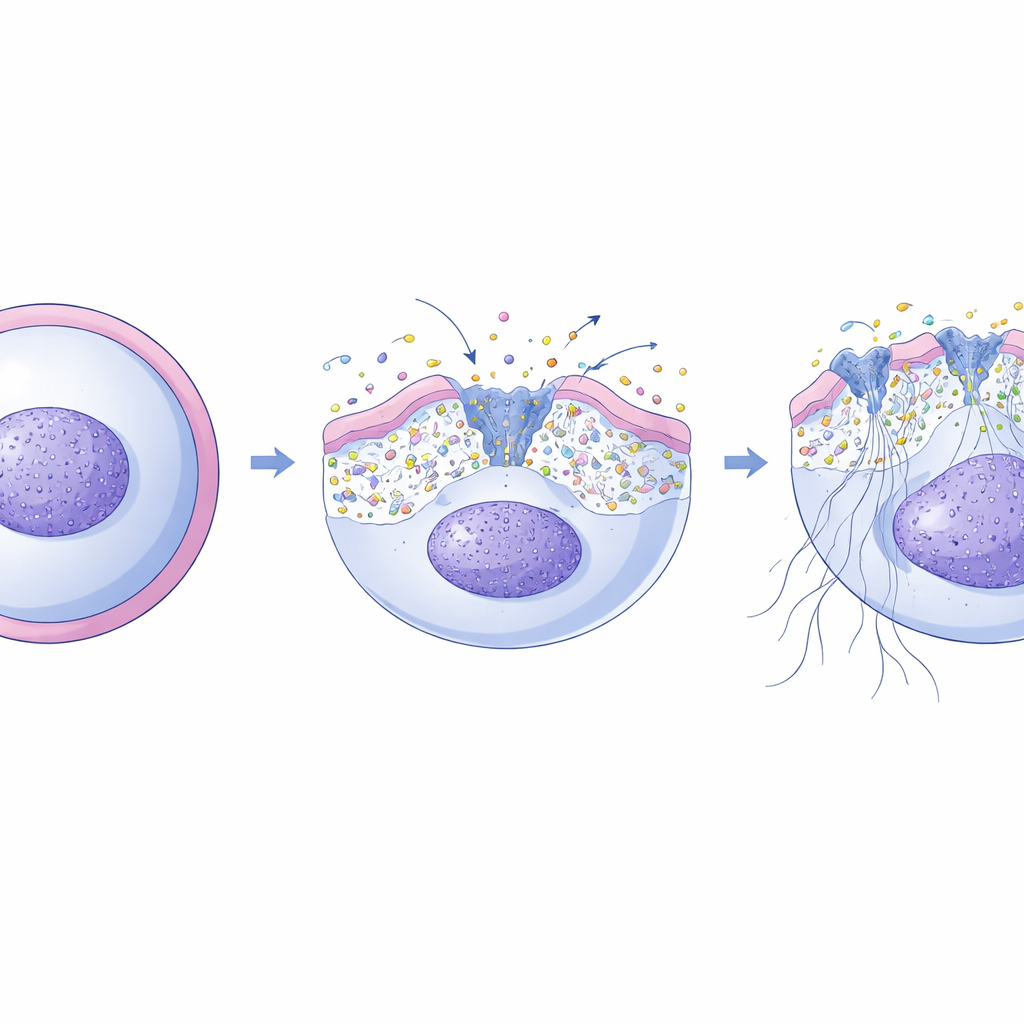
Das Team konzentrierte sich auf ein Protein namens XKR8, das besonders hoch in Neutrophilen vorkommt. XKR8 ist eine „Scramblase“ – ein Protein, das Lipide zwischen der inneren und äußeren Schicht der Zellmembran umverteilen kann und damit die übliche Asymmetrie zwischen den beiden Seiten aufhebt. In anderen Zusammenhängen, etwa beim programmierten Zelltod, ist bekannt, dass XKR8 ein Lipidsignal an der Zelloberfläche freilegt, das sterbende Zellen zur Entfernung markiert. Hier zeigten die Autoren, dass bei der NET-Bildung die Freilegung dieses Lipidsignals dem Membranleck und der DNA-Freisetzung vorausgeht und jene Neutrophilen markiert, die NETs bilden werden. Entfernten sie XKR8 aus Maus- und Humanneutrophilen, konnten die Zellen diesen frühen Scrambling-Schritt nicht mehr ausführen und versagten nahezu vollständig bei der NET-Bildung als Reaktion auf viele chemische und mikrobielle Stimuli.
Von Membran-Umschichtung zur Calciumflut
Bekannt ist, dass die NET-Bildung von einem Anstieg des intrazellulären Calciums in Neutrophilen abhängt, was Enzyme aktiviert, die Chromatin lockern und das Austreten von DNA ermöglichen. Die Forscher entdeckten, dass XKR8 im Zentrum dieses Prozesses steht. Nach einem oxidativen Burst innerhalb des Neutrophils schneidet ein „Vollstrecker“-Enzym, Caspase-3, XKR8 und aktiviert es. Einmal eingeschaltet, verteilt XKR8 schnell mehrere Lipidtypen über die Membran und nicht nur ein einzelnes „Fress mich“-Signal. Mit fluoreszenten Sonden zeigte das Team, dass diese Umschichtung die physikalische Spannung der Membran verändert. Diese mechanische Spannung öffnet daraufhin eine Reihe mechanosensitiver Calciumkanäle in der Membran, die Calciumionen genau in dem Zeitfenster einströmen lassen, in dem die NET-Bildung abläuft. Blockierten die Forscher diese Kanäle, gingen NETs größtenteils verloren; stimulierten sie die Kanäle mit speziellen Aktivatoren, konnten sie die NET-Bildung sogar in Zellen ohne XKR8 wiederherstellen.
Wenn Fallen versagen: Lungenschädigung und Pilzinfektion
Um zu verstehen, was dieser Membranschalter in lebenden Tieren bedeutet, untersuchten die Forscher Mäuse, denen entweder XKR8 vollständig fehlte oder die eine mutierte Version trugen, die nicht von Caspase-3 geschnitten werden kann. In Modellen akuter Lungenschädigung, ausgelöst durch bakterielle Komponenten, entwickelten normale Mäuse reichlich NETs in der Lunge, hohe Mengen freier DNA in der Lungenflüssigkeit und ausgeprägte Gewebeschäden. Mäuse mit eingeschränkter XKR8-Aktivität rekrutierten zwar die gleiche Zahl an Neutrophilen, produzierten aber deutlich weniger NETs und erlitten weniger Lungenschäden, was darauf hindeutet, dass der XKR8-Schalter ein wesentlicher Treiber schädlicher NET-Freisetzung in diesem Kontext ist. Im Gegensatz dazu waren Mäuse ohne funktionierenden XKR8-Schalter bei Lungeninfektionen mit dem Pilz Candida albicans schwer benachteiligt: Sie bildeten weit weniger NETs, trugen höhere Pilzlasten in Lunge und anderen Organen, bluteten stärker in die Lungenräume und starben häufiger an der überwältigenden Infektion. Bemerkenswert ist, dass ein Medikament, das einen der mechanosensitiven Calciumkanäle aktiviert, die Pilzbekämpfung bei Mäusen mit XKR8-defizienten Neutrophilen teilweise wiederherstellte.
Ein zentraler Hebel mit zwei Gesichtern
Diese Arbeit enthüllt einen vereinheitlichenden Mechanismus, durch den viele verschiedene Gefahrensignale auf einen einzigen membranbasierten Entscheidungspunkt in Neutrophilen zulaufen. Oxidative Bursts führen zur Aktivierung von Caspase-3, die XKR8 einschaltet, wodurch Membranlipide umgeschichtet, die Membranspannung verändert und Calciumkanäle geöffnet werden, was schließlich zur Auslösung von NETs führt. Für Laien lautet die Botschaft: Unser Immunsystem nutzt einen fein abgestimmten „Lipid-Hebel“ auf der Oberfläche von Neutrophilen, um zu entscheiden, wann DNA-Netze ausgeworfen werden. Ein Kippen dieses Hebels in die eine Richtung hilft, große Pilzangreifer zu beseitigen; ein zu häufiges oder fehlkoordiniertes Kippen kann sterile Entzündungen wie Lungenschäden oder Arthritis verschlimmern. Da dieser Weg an der Schwelle von Schutz und Schaden liegt, könnte das Anvisieren der Caspase-3–XKR8–Calcium-Achse neue Strategien bieten, um schädliche NETs zu dämpfen oder nützliche NETs zu stärken, je nach klinischem Bedarf.
Zitation: Liu, W., Ping, J., Deng, L. et al. Lipid asymmetry disruption by XKR8 orchestrates neutrophil extracellular trap formation and inhibits fungal infection. Nat Immunol 27, 949–960 (2026). https://doi.org/10.1038/s41590-026-02456-z
Schlüsselwörter: neutrophile extrazelluläre Netze, XKR8-Scramblase, angeborene Immunität, Pilz-Lungeninfektion, akute Lungenschädigung