Clear Sky Science · en
TSP-1 interaction with RANK and OPG: implications for bone remodeling and osteolytic bone metastasis
Why Bone-Breaking Cancers Matter
When cancers such as breast cancer spread to the skeleton, they can trigger painful “holes” in bone that lead to fractures, weakness, and a poorer quality of life. These lesions arise because cells that normally reshape bone become overactive and chew away too much tissue. The study summarized here uncovers a natural brake within this system: a fragment of a common connective‑tissue protein that can dial down bone destruction and slow cancer‑driven bone damage in mice.
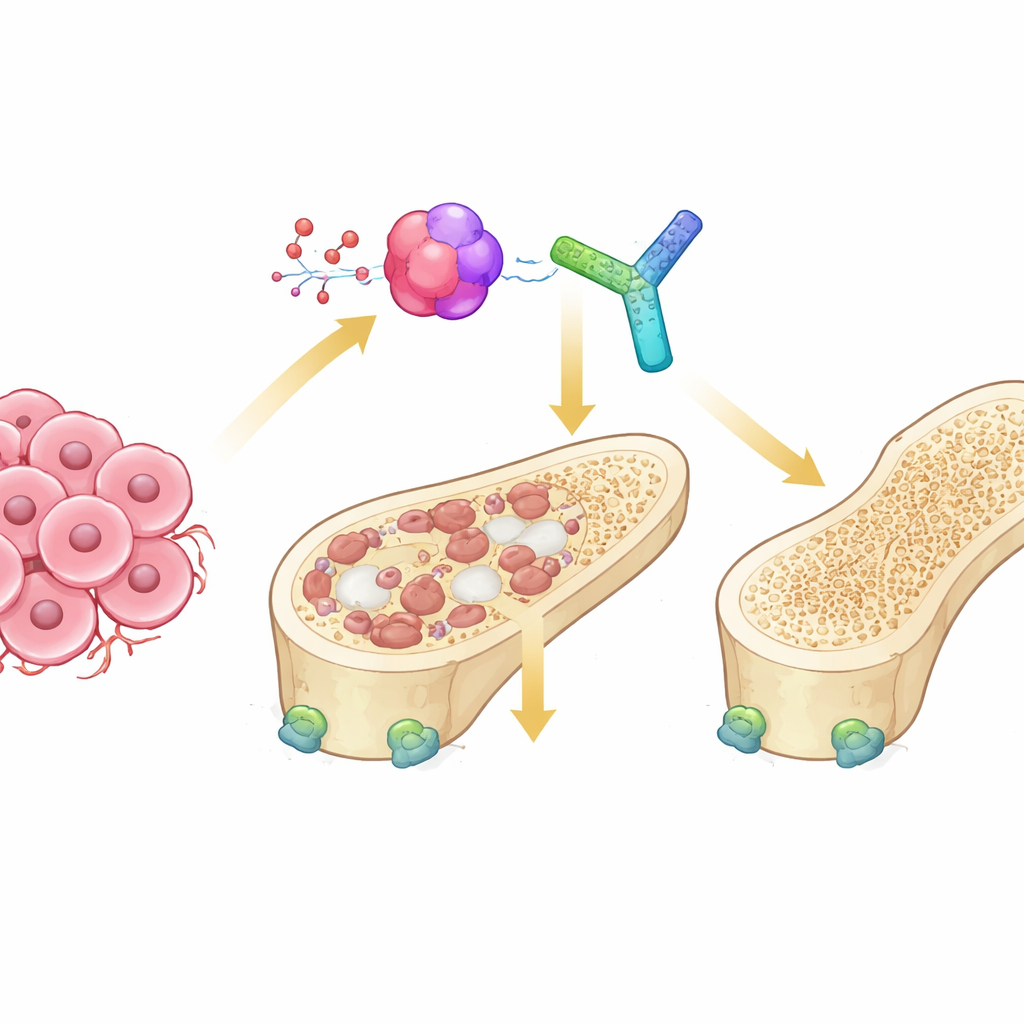
The Tug‑of‑War Inside Our Bones
Healthy bones are constantly renewed by two opposing cell types. Osteoclasts break down old bone, while osteoblasts build new bone. In cancers that spread to bone, this balance is lost: tumor cells send signals that overstimulate osteoclasts, leading to “osteolytic” lesions where bone is hollowed out. A key communication line in this process is the RANK–RANKL system, which switches on the maturation of osteoclasts. A third partner, osteoprotegerin (OPG), normally acts as a safety catch, binding RANKL and preventing it from overactivating bone‑eating cells. How this trio is fine‑tuned in the harsh environment of bone metastases has been unclear.
A Surprising Helper Hidden in a Larger Protein
The researchers focused on thrombospondin‑1 (TSP‑1), a large extracellular protein known to shape tissue architecture and cancer microenvironments. Previous work suggested full‑length TSP‑1 can actually encourage bone breakdown under some conditions. Here, the team examined a specific C‑terminal portion of TSP‑1, called E123CaG, which lacks some of the domains that stimulate osteoclasts. In cell cultures, they found that while intact TSP‑1 did not change RANKL‑induced osteoclast formation, the E123CaG fragment strongly reduced the number and size of mature osteoclasts and disrupted the actin ring structure those cells need to resorb bone. Importantly, mature osteoclasts themselves release proteases, especially an enzyme named HTRA1, that cut TSP‑1 and generate a fragment similar to E123CaG—hinting that this inhibitor may be produced as a built‑in feedback control.
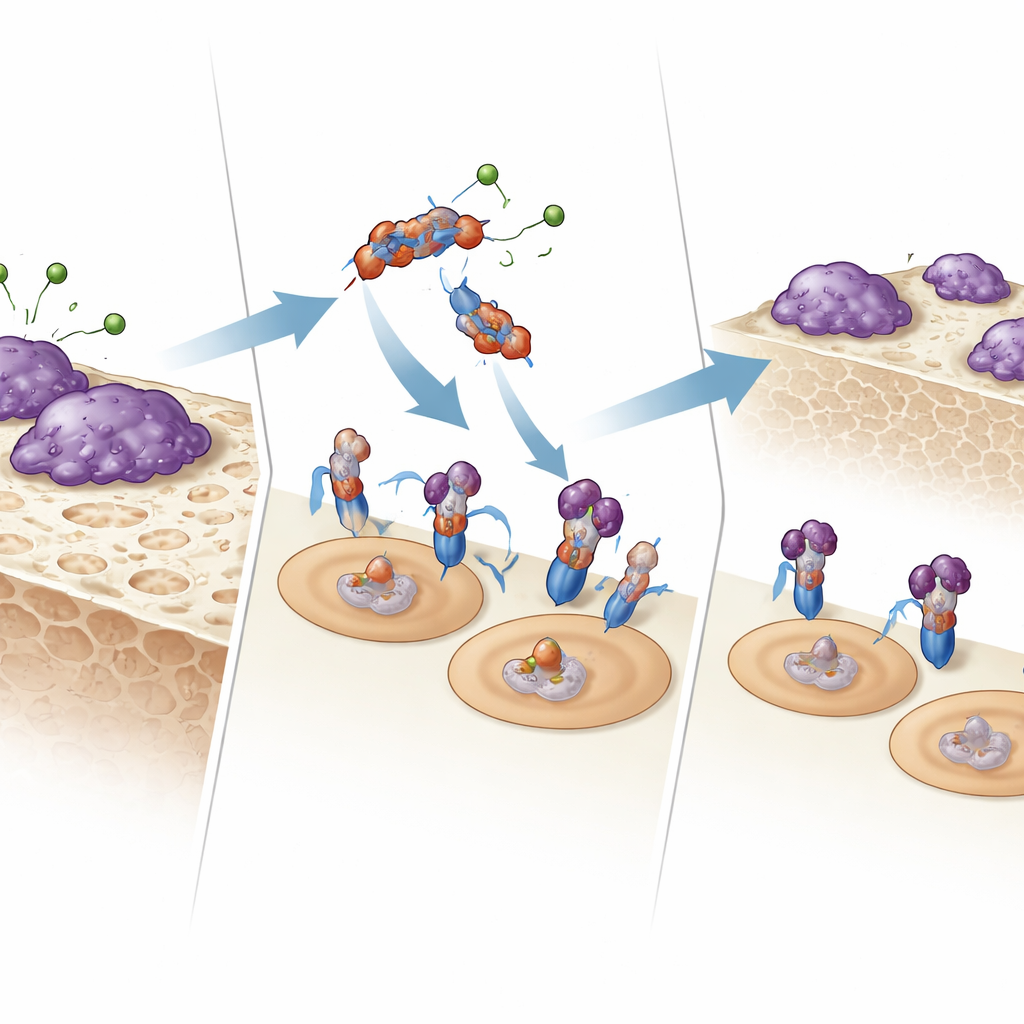
How the Fragment Talks to Bone‑Eating Cells
To understand how E123CaG works, the team tested whether it binds directly to the receptors that control osteoclasts. Using surface plasmon resonance and solid‑phase binding assays, they showed that E123CaG attaches to RANK, the receptor on osteoclast precursors that senses RANKL. This binding does not block RANKL from docking, but it does weaken the downstream signal: early signaling proteins (MAPKs p38 and JNK) turn on less strongly, and the key transcription factor NFATc1 moves into the nucleus less efficiently. Together, these changes blunt the maturation program that normally converts precursors into fully active bone‑resorbing cells.
Protecting the Natural Brake on Bone Loss
The fragment does more than interfere with RANK signaling. It also binds OPG, the body’s own decoy receptor that mops up RANKL. In culture, E123CaG did not stop OPG from capturing RANKL; instead, it boosted OPG’s ability to inhibit osteoclast formation. The researchers discovered why: during osteoclast development, proteases from mature osteoclasts, including HTRA1, chop up and inactivate OPG. When E123CaG was present, OPG was shielded from this degradation, remaining intact for longer. Thus, the same fragment generated by proteolysis of TSP‑1 both dampens RANK’s internal signal and preserves OPG, reinforcing two layers of control over bone destruction.
Testing the Fragment in a Living System
To see if these effects matter in a whole organism, the team engineered a mouse breast cancer cell line that preferentially spreads to bone to secrete the E123CaG fragment. When these cells were injected into the bloodstream of mice to seed bone metastases, animals harboring E123CaG‑producing tumors survived longer and had less severe bone damage than controls. High‑resolution micro‑CT scans showed better preservation of trabecular (spongy) bone, thicker struts, and smaller gaps between them. These benefits occurred without changes in the primary tumor’s growth rate, suggesting that the fragment mainly acts by modifying the bone microenvironment rather than directly attacking cancer cells.
What This Means for Patients
Taken together, the findings reveal a previously unrecognized protective circuit in bone: osteoclasts release proteases that clip TSP‑1 into a fragment which, in turn, reins in further osteoclast activation and shields the body’s natural inhibitor OPG from destruction. In the context of breast cancer bone metastasis, this fragment can slow the formation of bone‑eating cells and limit the spread of destructive lesions in mice. While more work is needed before any therapy can be developed, the study highlights E123CaG and its interactions with RANK and OPG as promising leads for drugs that could better protect bones in cancer and other diseases marked by excessive bone loss.
Citation: Carminati, L., Sangalli, F., Urbinati, C. et al. TSP-1 interaction with RANK and OPG: implications for bone remodeling and osteolytic bone metastasis. Cell Death Dis 17, 332 (2026). https://doi.org/10.1038/s41419-026-08600-9
Keywords: bone metastasis, osteoclasts, thrombospondin-1, RANKL RANK OPG, bone remodeling