Clear Sky Science · en
Androgen receptor and fatty acid oxidation cooperate in ferroptosis evasion in BRAFi resistant melanoma
Why this matters for treating stubborn skin cancers
Drugs that block mutant BRAF, a key growth switch in melanoma, have transformed care for people with advanced skin cancer. Yet many tumors initially shrink only to come roaring back months later. This study digs into what helps those comeback tumors stay alive and explores a new way to push them into a self-destruct mode called ferroptosis, using two existing heart and prostate drugs in combination.
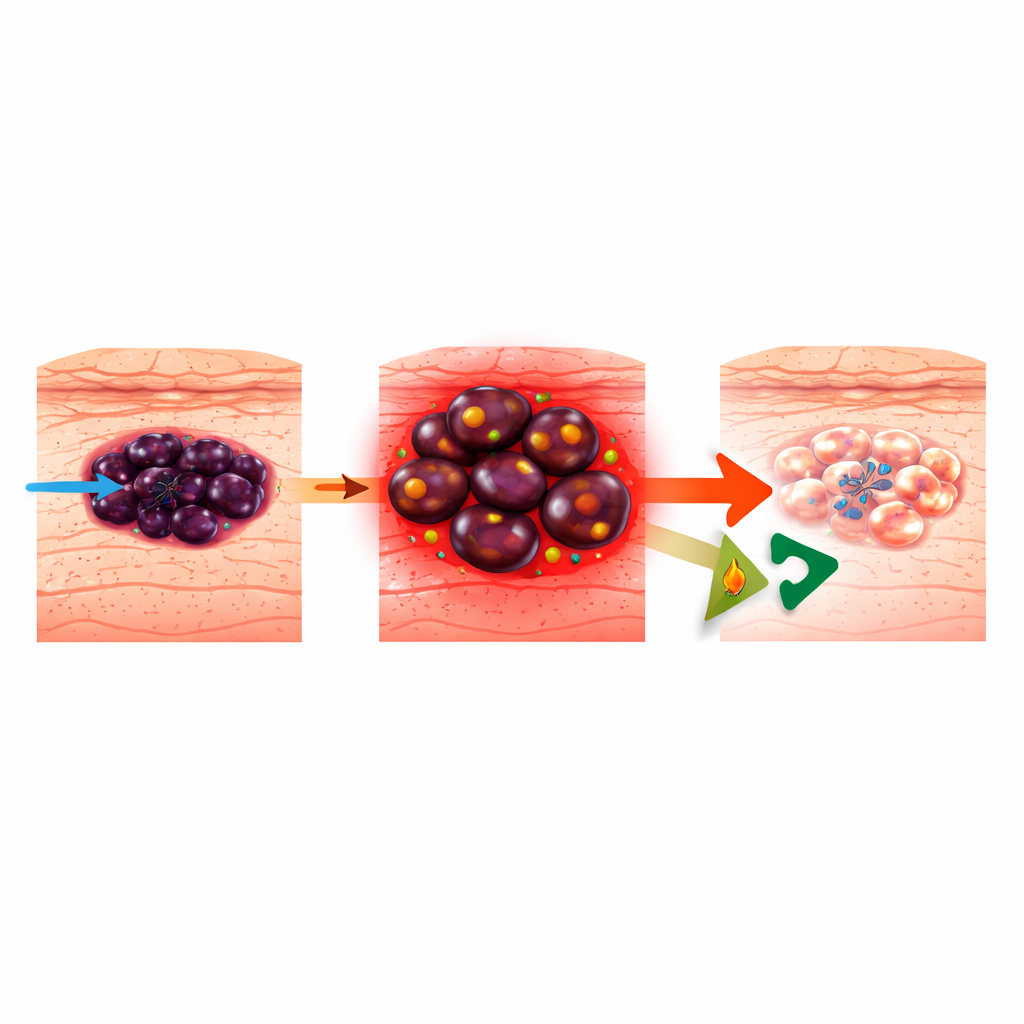
How melanoma learns to dodge targeted drugs
Melanoma arises from pigment-producing cells in the skin and is responsible for most skin cancer deaths. Modern targeted therapies go after the overactive BRAF–MEK pathway that drives many melanomas, but tumors are remarkably adaptable. They can switch between different “cell states,” from a more mature pigment-making identity to a more primitive, neural-crest–like or undifferentiated state that is aggressive and drug-resistant. Earlier work showed that as melanomas become resistant to BRAF-blocking drugs (BRAFi), they increasingly fuel themselves by burning fats in their mitochondria, a process called fatty acid oxidation (FAO). Blocking FAO with the heart drug ranolazine was known to slow the rise of resistant cells, but how this actually killed melanoma cells was unclear.
A hidden vulnerability: iron-driven fat damage
The researchers focused on ferroptosis, a form of cell death triggered when iron helps generate destructive chemical reactions in certain fats within cell membranes. They found that BRAFi-resistant melanoma cells carry many features that leave them on the brink of ferroptosis: they have lower levels of the antioxidant glutathione, pack their membranes with long, damage-prone polyunsaturated fats, and show more oxidized membrane lipids than their drug-sensitive counterparts. Patient tumor samples that had progressed on BRAF inhibitors showed the same pattern, with markers of both ferroptosis and fat-burning pathways rising together. This suggested that resistant cells walk a tightrope—highly vulnerable to iron-driven fat damage, yet simultaneously wired to suppress it.
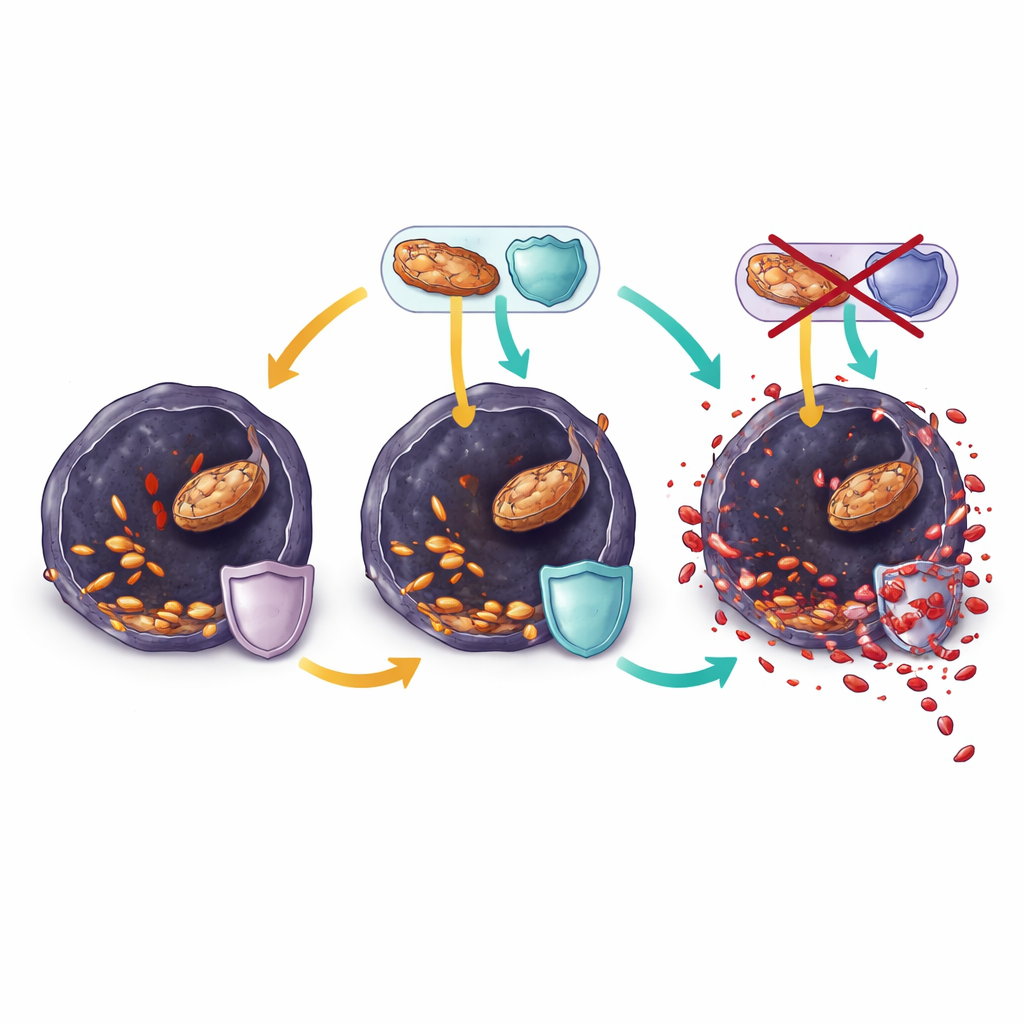
Ranolazine tips the balance toward cell death
In resistant melanoma cells grown in the lab, ranolazine rapidly cut down fat-burning activity, drained key energy molecules, and further lowered glutathione. At the same time, more polyunsaturated fats were stuffed into membrane lipids, and signs of oxidative stress and lipid damage surged. These changes slowed cell growth within hours and could be reversed by classic ferroptosis blockers, showing that ranolazine was killing cells mainly by pushing them over the edge into ferroptosis. However, when tumors in mice eventually escaped combined BRAF and ranolazine treatment, their cells had rewired again. They maintained high levels of enzymes that repair oxidized lipids and replenish protective molecules, and they remodeled their membranes to contain more resistant monounsaturated fats instead of vulnerable polyunsaturated ones.
Hormone signals help rebuild the tumor’s defenses
The team traced this membrane remodeling to enzymes called MBOAT1 and MBOAT2, which swap in safer fats during phospholipid renewal. These enzymes were strongly increased in ranolazine-resistant cells and in many patient tumors that had relapsed on BRAF inhibitors. Their activity depended on the androgen receptor (AR), a hormone sensor better known from prostate cancer. AR bound to control regions of the MBOAT1 and MBOAT2 genes and boosted their activity. When the researchers blocked AR with the prostate cancer drug enzalutamide, levels of MBOAT1/2 dropped and melanoma cells became much more sensitive to ferroptosis-inducing agents. This effect was seen across different melanoma cell states, including both primitive, highly drug-resistant cells and more differentiated pigment-like cells.
Pairing two old drugs to outsmart resistant tumors
Because FAO and AR-controlled lipid remodeling each help melanoma cells escape ferroptosis, the authors tested whether hitting both at once would be particularly effective. In cell culture, combining ranolazine (which blocks fat burning and boosts lipid damage) with enzalutamide (which disarms AR and its membrane-protective enzymes) caused extensive ferroptotic cell death, especially in cells that relied heavily on FAO and had shifted into the aggressive undifferentiated state. Under conditions that force melanoma cells to depend more on fat as fuel, this combination became even more deadly. The work suggests that a dual-therapy strategy using these already approved drugs could both delay resistance to BRAF-targeted treatments and make resistant tumors, including those that blunt immune attack, more vulnerable by forcing them into ferroptosis.
What this could mean for patients
This research paints a detailed picture of how melanoma cells, driven into a corner by targeted therapy, reshape their metabolism and hormone signaling to dodge a lethal form of iron- and fat-based cell death. By revealing that fat-burning pathways and the androgen receptor cooperate to keep ferroptosis in check, it points to a practical way to pull out that safety net. While clinical testing will be required, the idea of repurposing heart and prostate drugs alongside existing melanoma treatments offers a promising path to longer-lasting responses and potentially better outcomes for patients whose tumors are prone to relapse.
Citation: Redondo-Muñoz, M., Caballe-Mestres, A., Reisz, J.A. et al. Androgen receptor and fatty acid oxidation cooperate in ferroptosis evasion in BRAFi resistant melanoma. Cell Death Dis 17, 338 (2026). https://doi.org/10.1038/s41419-026-08578-4
Keywords: melanoma, ferroptosis, fatty acid oxidation, androgen receptor, drug resistance