Clear Sky Science · de
Die katalytische enantioselektive [1,2]-Wittig-Umlagerungs-Kaskade von allylischen Ethern
Aus molekularem Durcheinander Ordnung schaffen
Chemiker wollen häufig Moleküle herstellen, die spiegelbildselektiv sind, weil unsere Körper „linkshändige“ und „rechtshändige“ Formen unterscheiden können. Dieser Artikel behandelt ein langjähriges Rätsel in diesem Bereich: Wie kann man eine notorisch unruhige Art von Bindungsverschiebungsreaktion so lenken, dass sie überwiegend eine Spiegelbildform liefert statt eines zufälligen Gemischs. Die Autorinnen und Autoren finden nicht nur einen Weg dazu, sie zeigen außerdem, dass die Reaktion über eine unerwartete Route verläuft, die Jahrzehnte von Lehrbuchwissen in Frage stellt.
Warum diese Formveränderungen wichtig sind
Viele leistungsfähige Reaktionen in der organischen Chemie funktionieren, indem sie Bindungen innerhalb eines Moleküls verschieben, anstatt alles aufzubrechen und neu zu beginnen. Solche „Umlagerungen“ sind geschätzt, weil sie wenig Material verschwenden und komplexe Strukturen in nur einem Schritt aufbauen können. Zu diesen Reaktionen gehören Wittig‑Umlagerungen, die einfache Ethergruppen in wertvolle Alkohole verwandeln können. Eine spezielle Variante, die sogenannte [1,2]-Wittig‑Umlagerung von allylischen Ethern, war jedoch schwer zu kontrollieren: Sie liefert typischerweise Produktgemische und verwischt entscheidend die 3‑dimensionale Anordnung der Atome, statt eine einzige Händigkeit zu bewahren oder zu erzeugen.
Ein neuer Kaskadenweg
Die Forscher zeigen, dass sich diese problematische Reaktion zähmen lässt, wenn man sie als Teil einer zweiteiligen Kaskade ausführt, die von einem speziell entwickelten organischen Katalysator geleitet wird. Ihr Katalysator, eine bifunktionelle Iminophosphoran‑Verbindung, ist sowohl eine außergewöhnlich starke Base als auch eine präzise „handige“ Umgebung. Zuerst löst er eine gut gesteuerte Bindungsverschiebung aus, bekannt als [2,3]-Umlagerung, die ein chirales tertiäres Alkohol mit sehr hoher Präferenz für eine Enantiomer bildet. Unter basischen Bedingungen rearrangiert dieses Zwischenprodukt dann stillschweigend weiter zum finalen [1,2]-Wittig‑Produkt. Über viele verschiedene Ausgangsstoffe liefert die Sequenz zuverlässig chirale homoallylische Alkohole in guten Ausbeuten und mit hohen Enantiomerenverhältnissen, das heißt eine Händigkeit dominiert deutlich.
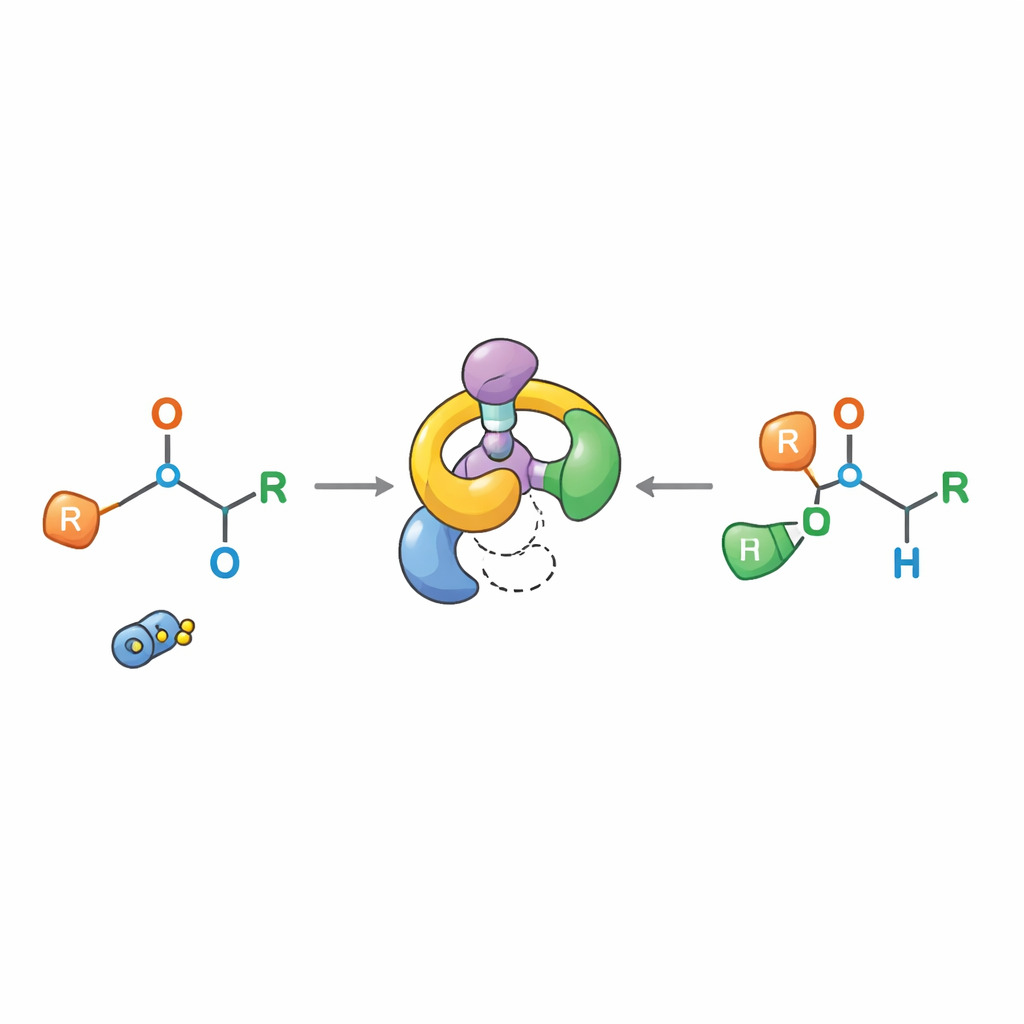
Untersuchungen, wie die Reaktion wirklich abläuft
Traditionelle Erklärungen gingen davon aus, dass [1,2]-Wittig‑Produkte durch Bindungsbruch entstehen, der ein lockeres Radikalpaar bildet, das später wieder rekombiniert — ein Bild, das eine präzise stereochemische Kontrolle unwahrscheinlich erscheinen ließ. Die neue Arbeit zeichnet ein anderes Bild. Durch sorgfältige Experimente — darunter Fangversuche mit radikal‑empfindlichen Additiven, „Crossover“-Tests, die verschiedene Moleküle mischen, und detaillierte zeitaufgelöste NMR‑Messungen — zeigen die Autorinnen und Autoren, dass die Reaktion über ein spezifisches [2,3]-umgelagertes Zwischenprodukt verläuft und dass Atome innerhalb eines einzelnen Moleküls gepaart bleiben, statt die Partner zu tauschen. Der zweite Schritt verhält sich formal wie eine verbotene [1,3]-Umlagerung, in der Praxis läuft er jedoch über Fragmentierung in ein eng assoziiertes Ionenpaar ab, das hochgeordnet wieder zusammenklappt und so die Konfiguration am chiralen Zentrum erhält.
Computergestützte Bestätigung der verborgenen Schritte
Um dieses mechanistische Bild zu untermauern, führte das Team umfangreiche quantenchemische Berechnungen an realistischen Modellen von Katalysator und Substraten durch. Diese Rechnungen zeigen, warum in dem ersten Schritt ein Enantiomer bevorzugt wird: Der bevorzugte Übergangszustand bildet ein Netzwerk aus drei starken Wasserstoffbrücken und eine günstige Stapelwechselwirkung zwischen aromatischen Ringen, während konkurrierende Anordnungen unter schwächeren Wechselwirkungen und sterischer Behinderung leiden. Für den zweiten Schritt finden die Berechnungen keinen einzigen glatten, konzertierten Weg; stattdessen stützen sie einen schrittweisen ionischen Fragmentierungsweg mit einer mäßigen Energiebarriere, gefolgt von nahezu barrierefreier Rekombination. Die prognostizierte Energiebarriere stimmt eng mit den experimentell gemessenen Werten überein und stärkt damit das Argument für den Ionenpaar‑Mechanismus.
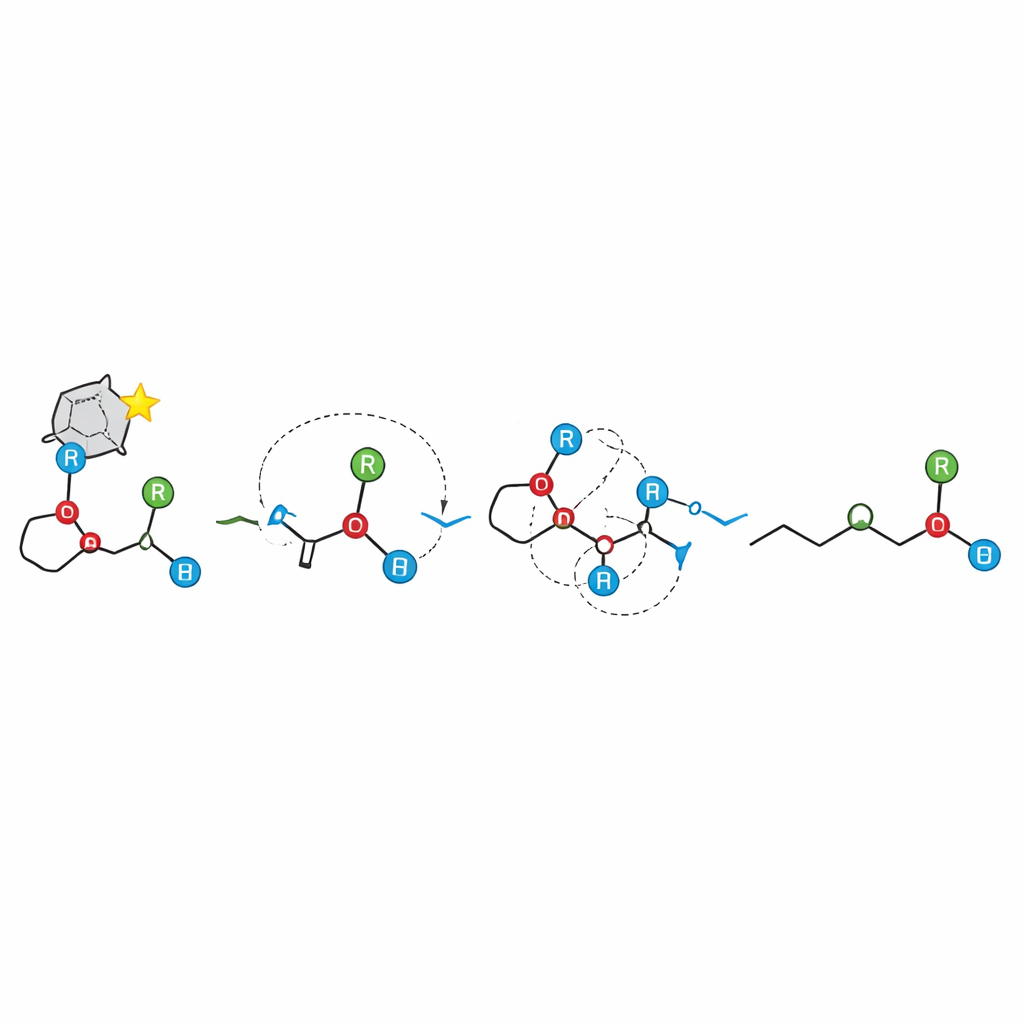
Folgen für den Aufbau chiraler Moleküle
Praktisch gesehen liefert diese Arbeit synthetischen Chemikern ein robustes neues Werkzeug, um chirale tertiäre Alkohole aus leicht verfügbaren allylischen Ethern herzustellen, und sie zeigt, welche strukturellen Veränderungen den Prozess begünstigen oder behindern. Allgemeiner demonstriert sie, dass stereochemische Information selbst dann mit bemerkenswerter Treue übertragen werden kann, wenn eine Reaktion über geladene Fragmente verläuft, solange diese Fragmente eng assoziiert bleiben. Indem die Studie die radikalbasierte Sicht der [1,2]-Wittig‑Umlagerung revidiert und eine hochselektive ionische Kaskade vorstellt, eröffnet sie die Möglichkeit, andere komplexe Bindungsumlagerungen neu zu überdenken und für präzise dreidimensionale Kontrolle umzugestalten.
Zitation: Kang, T., O’Yang, J., Kasten, K. et al. The catalytic enantioselective [1,2]-Wittig rearrangement cascade of allylic ethers. Nat. Chem. 18, 800–809 (2026). https://doi.org/10.1038/s41557-025-02022-4
Schlüsselwörter: asymmetrische Katalyse, sigmatrope Umlagerung, chirale tertiäre Alkohole, Reaktionsmechanismus, organische Synthese